ビリアード錠300mgの添付文書
- 商品名:
- ビリアード錠300mg
- 一般名:
- テノホビル ジソプロキシルフマル酸塩
- 略称 :
- TDF

添付文書の読み方
ここで提供している添付文書情報は、2024年4月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
抗ウイルス化学療法剤
テノホビル ジソプロキシルフマル酸塩
- **2023年8月改訂(第3版)
- *2021年6月改訂(第2版)
- 劇薬
処方箋医薬品注)
注)注意-医師等の処方箋により使用すること
| 日本標準商品分類番号 | 87625 |
|---|
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 48箇月 |
| 承認番号 | 21600AMY00073000 |
|---|---|
| 販売開始 | 2004年4月 |
1. 警告
B型慢性肝炎を合併している患者では,本剤の投与中止により,B型慢性肝炎が再燃するおそれがあるので,本剤の投与を中断する場合には十分注意すること。特に非代償性の場合,重症化するおそれがあるので注意すること。[9.1.1 参照]
2. 禁忌(次の患者には投与しないこと)
本剤の成分に対し過敏症の既往歴のある患者
3. 組成・性状
3.1 組成
| 有効成分 1錠中 |
テノホビル ジソプロキシルフマル酸塩300mg (テノホビル ジソプロキシルとして245mg) |
|---|---|
| 添加剤 | クロスカルメロースNa,乳糖,ステアリン酸Mg,セルロース,部分アルファー化デンプン,青色2号,ヒプロメロース,酸化チタン,トリアセチン |
3.2 製剤の性状
| 剤形 | フィルムコーティング錠 | |
|---|---|---|
| 性状 | うすい青色 | |
| 外形 | 上面 |  |
| 下面 |  |
|
| 側面 |  |
|
| 大きさ | 長径 | 約17.0mm |
| 短径 | 約10.5mm | |
| 厚さ | 約5.0mm | |
| 識別コード | GILEAD4331-300 | |
4. 効能又は効果
HIV-1感染症
6. 用法及び用量
通常,成人にはテノホビル ジソプロキシルフマル酸塩として1回300mg(テノホビル ジソプロキシルとして245mg)を1日1回経口投与する。なお,投与に際しては必ず他の抗HIV薬と併用すること。
7. 用法及び用量に関連する注意
7.1
腎機能障害のある患者では本剤の血中濃度が上昇するので,腎機能の低下に応じて,次の投与方法を目安とする。[8.3 参照],[9.2.1 参照],[10.2 参照],[11.1.1 参照],[16.6.1 参照]
| クレアチニン |
投与方法 |
|---|---|
| 50mL/min以上 | 本剤1錠を1日1回投与 |
| 30~49mL/min | 本剤1錠を2日間に1回投与 |
| 10~29mL/min | 本剤1錠を1週間に2回投与 |
| 血液透析患者 | 本剤1錠を1週間に1回投与注)又は累積約12時間の透析終了後に本剤1錠を投与 |
注) 血液透析実施後
7.2
本剤の有効成分であるテノホビル ジソプロキシルフマル酸塩を含む製剤と併用しないこと。また,テノホビル アラフェナミドフマル酸塩を含む製剤についても併用しないこと。
7.3
核酸系逆転写酵素阻害薬(NRTI)3成分のみを用いる一部の治療は,NRTI2成分に非核酸系逆転写酵素阻害薬又はHIV-1プロテアーゼ阻害薬を併用する3成分併用療法と比べて,概して効果が低いことが報告されている。また,抗ウイルス薬の使用経験がない患者に対し,本剤とジダノシン,ラミブジン又は本剤とラミブジン,アバカビルの3剤併用1日1回投与により,初期のウイルス学的応答の欠如が高頻度に認められたとの報告があるので,抗ウイルス薬の使用経験がない患者及び既治療患者に対して本剤を使用する場合には,これらの組み合わせのみによる治療は避けること。
8. 重要な基本的注意
**,*8.1
本剤の使用に際しては,国内外のガイドライン等の最新の情報を参考に,患者又はそれに代わる適切な者に次の事項についてよく説明し同意を得た後,使用すること。
8.1.1
本剤はHIV感染症の根治療法薬ではないことから,日和見感染症を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので,本剤投与開始後の身体状況の変化についてはすべて担当医に報告すること。
8.1.2
本剤の長期投与による影響については現在のところ不明であること。
8.2
本剤を含む抗HIV薬の多剤併用療法を行った患者で,免疫再構築炎症反応症候群が報告されている。投与開始後,免疫機能が回復し,症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス,サイトメガロウイルス,ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また,免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症,多発性筋炎,ギラン・バレー症候群,ブドウ膜炎等)が発現するとの報告があるので,これらの症状を評価し,必要時には適切な治療を考慮すること。
8.3
本剤投与前にクレアチニンクリアランス,尿糖及び尿蛋白の検査を実施すること。また,本剤投与後も定期的な検査等により患者の状態を注意深く観察すること。[7.1 参照],[9.2.1 参照],[10.2 参照],[11.1.1 参照],[16.6.1 参照]
8.4
テノホビル ジソプロキシルフマル酸塩を含む多剤併用療法を長期間行った患者において,骨粗鬆症が現れ,大腿骨頚部等の骨折を起こした症例が報告されている。長期投与時には定期的に骨密度検査を行う等骨密度減少に注意し,異常が認められた場合には適切な処置を行うこと。なお,本剤の試験において,144週間の投与により腰椎と大腿骨頚部の骨密度の減少が見られている。骨密度の減少した患者の大部分は,投与開始後24~48週目にかけて発現し,以降は144週目まで持続していた。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 B型肝炎ウイルス感染を合併している患者
本剤の投与を中断する場合には十分注意すること。B型慢性肝炎を合併している患者では,本剤の投与中止により,B型慢性肝炎が再燃するおそれがある。特に非代償性の場合,重症化するおそれがある。[1. 参照]
9.1.2 腎機能障害のリスクを有する患者
血清リンの検査を実施すること。
9.2 腎機能障害患者
9.2.1 中等度及び重度の腎機能障害のある患者
本剤の血中濃度が上昇する。[7.1 参照],[8.3 参照],[10.2 参照],[11.1.1 参照],[16.6.1 参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には,治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物試験(サル)においてテノホビルの胎児への移行が報告されている1)。
9.6 授乳婦
授乳を避けさせること。テノホビルのヒト乳汁への移行が報告されており2),動物実験(ラット)において,乳汁中への移行が報告されている。また,女性のHIV感染症患者は,乳児のHIV感染を避けるため,乳児に母乳を与えないことが望ましい。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の肝,腎及び心機能の低下,合併症,併用薬等を十分に考慮すること。
10. 相互作用
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| 逆転写酵素阻害剤 ジダノシン [16.7 参照] |
ジダノシンによる有害事象を増強するおそれがあるので,ジダノシンの減量を考慮すること。 | ジダノシンのAUC及びCmaxが上昇する。 |
| HIVプロテアーゼ阻害剤 アタザナビル硫酸塩 [16.7 参照] |
アタザナビルの治療効果が減弱するおそれがあるので,本剤とアタザナビル硫酸塩を併用する場合には,本剤とアタザナビル300mgをリトナビル100mgとともに投与することが望ましい。また,本剤による有害事象を増強するおそれがある。 | アタザナビルのAUCが25%,Cmaxが21%,Cminが40%低下し,テノホビルのAUCが24%,Cmaxが14%,Cminが22%上昇する。 |
| HIVプロテアーゼ阻害剤 ロピナビル・リトナビル [16.7 参照] |
本剤による有害事象を増強するおそれがある。 | テノホビルのAUCが32%,Cminが51%上昇する。 |
| HIVプロテアーゼ阻害剤 ダルナビル+リトナビル [16.7 参照] |
本剤による有害事象を増強するおそれがある。 | テノホビルのAUC,Cmax及びCminが上昇する。 |
| 抗HCV剤 レジパスビル・ソホスブビル [16.7 参照] |
本剤による有害事象を増強するおそれがある。 | テノホビルのAUC,Cmax及びCminが上昇する。 |
| 抗ウイルス化学療法剤 アシクロビル,バラシクロビル塩酸塩 抗サイトメガロウイルス化学療法剤 ガンシクロビル,バルガンシクロビル塩酸塩等 |
これらの薬剤又は本剤による有害事象を増強するおそれがある。 | 尿細管への能動輸送により排泄される薬剤と併用する場合,排泄経路の競合により,排泄が遅延し,これらの薬剤又は本剤の血中濃度が上昇するおそれがある。 |
| 腎毒性を有する薬剤 [7.1 参照],[8.3 参照],[9.2.1 参照],[11.1.1 参照],[16.6.1 参照] |
併用は避けることが望ましい。 | 腎毒性を有する薬剤は腎機能障害の危険因子となる。 |
11. 副作用
次の副作用が現れることがあるので,観察を十分に行い,異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1 重大な副作用
11.1.1 腎不全又は重度の腎機能障害(1.2%)
腎機能不全,腎不全,急性腎障害,近位腎尿細管機能障害,ファンコニー症候群,急性腎尿細管壊死,腎性尿崩症又は腎炎等の重度の腎機能障害が現れることがあるので,臨床検査値に異常が認められた場合には,投与を中止する等,適切な処置を行うこと。特に腎機能障害の既往がある患者や腎毒性のある薬剤が投与されている患者では注意すること。[7.1 参照],[8.3 参照],[9.2.1 参照],[10.2 参照],[16.6.1 参照]
11.1.2 膵炎(0.2%)
血中アミラーゼ,リパーゼ,血中トリグリセリド等の検査値の上昇がみられた場合には,投与を中止する等,適切な処置を行うこと。
11.1.3 乳酸アシドーシス及び脂肪沈着による重度の肝腫大(脂肪肝)(頻度不明)
乳酸アシドーシス又は肝細胞毒性が疑われる臨床症状又は検査値異常(アミノトランスフェラーゼの急激な上昇等)が認められた場合には,本剤の投与を一時中止すること。特に肝疾患の危険因子を有する患者においては注意すること。類薬(ヌクレオシド系逆転写酵素阻害薬)の単独投与又はこれらの併用療法により,重篤な乳酸アシドーシス及び脂肪沈着による重度の肝腫大(脂肪肝)が,女性に多く報告されている。
11.2 その他の副作用
| 2%以上 | 2%未満 | 頻度不明注1) | |
|---|---|---|---|
| 代謝及び栄養障害 | 食欲減退(3.2%),体重減少(2.1%),体脂肪の再分布/蓄積(2.1%) | 高コレステロール血症,高脂血症 | 低リン酸血症,低カリウム血症,糖尿病,高尿酸血症 |
| 精神障害 | うつ病,睡眠障害,リビドー減退,神経過敏,不安 | ||
| 神経系障害 | 頭痛(5.6%),錯感覚(3.7%),浮動性めまい(3.4%) | 不眠症,末梢性ニューロパチー,味覚異常,異常な夢,傾眠,ニューロパチー,思考異常,振戦 | 感覚鈍麻 |
| 呼吸器,胸郭及び縦隔障害 | 気管支炎,鼻炎,咽頭炎 | 呼吸困難 | |
| 胃腸障害 | 悪心(10.5%),下痢(9.1%),腹痛(5.2%),嘔吐(4.4%),鼓腸(3.0%),消化不良(2.3%) | 口内乾燥,胃腸障害,便秘,アフタ性潰瘍,胃炎,おくび,腹部膨満 | |
| 肝胆道系障害 | 肝炎 | 脂肪肝,肝機能異常 | |
| 皮膚及び皮下組織障害 | 発疹(3.3%) | そう痒症,多汗症,脱毛症,湿疹,ざ瘡,皮膚乾燥,単純ヘルペス,皮膚良性新生物 | |
| 筋骨格系及び結合組織障害 | 骨障害(2.1%) | 筋肉痛,関節痛,背部痛,側腹部痛,筋痙攣 | 骨軟化症,ミオパチー |
| 一般・全身障害及び投与部位の状態 | 無力症(6.3%),疼痛(2.4%) | 倦怠感,胸痛,発熱,悪寒,末梢性浮腫 | |
| 臨床検査注2) | CK増加(12.3%),血中トリグリセリド増加(7.8%),血中アミラーゼ増加(7.5%),AST増加(5.1%),ALT増加(4.3%),好中球数減少(2.4%),尿糖(2.1%),血中ブドウ糖増加(2.0%) | 血中ビリルビン増加,血中リン減少,Al-P増加,血小板数減少 | リパーゼ増加,血尿,蛋白尿,血中クレアチニン増加,γ-GTP増加 |
| その他 | 頻尿,視覚異常,多尿 | アレルギー反応,高血圧 |
- 注1) 市販後の調査,自発報告等にて報告された副作用
- 注2) 臨床検査についてはグレード3及び4(NIAID分類)の臨床検査値異常
13. 過量投与
13.1 処置
本剤は血液透析により一部除去される。[16.6.1 参照]
15. その他の注意
15.2 非臨床試験に基づく情報
マウスを用いたがん原性試験(2年間)において,臨床用量におけるヒトの全身曝露量の16倍で雌に肝細胞腺腫が高頻度に発現したとの報告がある。
16. 薬物動態
16.1 血中濃度
16.1.1
日本人健康成人男性に本剤300mgを空腹時に経口投与した場合,本剤の活性成分であるテノホビルの血清中濃度は1.2±0.5時間後に最高値に達し,Cmax及びAUCはそれぞれ212±43ng/mL及び2,197±516ng・hr/mLであった。テノホビルの消失は二相性を示し,最終相の半減期は15.1±2.3時間であった3)。
16.1.2
外国人健康成人に本剤300mgを空腹時単回経口投与した場合,テノホビルの血清中濃度は1.0±0.4時間後に最高値に達し,Cmax及びAUCは,それぞれ296±90ng/mL及び2,287±685ng・hr/mLであった。テノホビルの薬物動態は,本剤の投与量が75~600mgの範囲において用量に比例し,また,反復投与による影響を受けなかった。
16.2 吸収
16.2.1 食事の影響
本剤を軽食とともに服用した場合の薬物動態は,空腹時投与に比較し有意な変動はなかったが,高脂肪食(約700~1,000kcal,40~50%が脂肪由来)摂取後に本剤を服用した場合には,テノホビルのAUC及びCmaxは,それぞれ約40%及び約14%上昇した。本剤300mgを1日1回食後反復投与した場合の,テノホビルのCmax及びAUCは,それぞれ326±119ng/mL及び3,324±1,370ng・hr/mLであった(外国人における成績)。
16.3 分布
テノホビル1.0mg/kg及び3.0mg/kgを静脈内投与後の定常状態での分布容積は,それぞれ1.3±0.6L/kg及び1.2±0.4L/kgであった。テノホビルのヒト血漿及び血清蛋白結合率(in vitro)は,0.01~25μg/mLのテノホビル濃度範囲においてそれぞれ0.7%未満及び7.2%未満であった(外国人における成績)注1)。
注1) 本剤の承認された1日用量は経口投与300mgである。
16.4 代謝
本薬は活性成分をテノホビルとするジエステル化プロドラッグであり,経口投与後,速やかにテノホビルに代謝され,その後細胞内でテノホビル二リン酸に代謝される。また,in vitro試験から,テノホビル ジソプロキシル及びテノホビルはいずれもチトクロームP450の基質ではないことが示されている(外国人における成績)。
16.5 排泄
本剤300mgを空腹時に経口投与した際,投与後48時間までのテノホビルの尿中排泄率は24±4%であり,CLrenalは287±64mL/minであった(日本人における成績)3)。本剤300mgを1日1回食後反復経口投与した際,投与量の32±10%(テノホビル換算)が24時間以内に尿中に回収された。また,テノホビルを静脈内投与した場合は,投与量の70~80%が72時間までに,テノホビルとして尿中に回収された。テノホビルは,糸球体濾過と尿細管への能動輸送により腎排泄される(外国人における成績)。
16.6 特定の背景を有する患者
16.6.1 腎不全患者(919試験)
腎機能障害を有する患者を対象に,本剤300mgを単回投与した場合,クレアチニンクリアランス(CLcr)が50mL/min未満の患者あるいは透析を必要とする末期腎不全患者において,テノホビルのCmax及びAUCが上昇した(外国人における成績)(表1)。[7.1 参照],[8.3 参照],[9.2.1 参照],[10.2 参照],[11.1.1 参照],[13.1 参照]
| CLcr (mL/min) |
例数 | Cmax (ng/mL) |
AUC (ng・hr/mL) |
CL/F (mL/min) |
CLrenal (mL/min) |
|---|---|---|---|---|---|
| >80 | 3 | 335.5±31.8 | 2,184.5±257.4 | 1,043.7±115.4 | 243.5±33.3 |
| 50-80 | 10 | 330.4±61.0 | 3,063.8±927.0 | 807.7±279.2 | 168.6±27.5 |
| 30-49 | 8 | 372.1±156.1 | 6,008.5±2,504.7 | 444.4±209.8 | 100.6±27.5 |
| <30 (12-28)注2) |
11 | 601.6±185.3 | 15,984.7±7,223.0 | 177.0±97.1 | 43.0±31.2 |
- 平均値±標準偏差
- 注2) CLcrが10mL/min未満で,透析を行っていない患者における薬物動態は検討されていない。
なお,血液透析による除去率は54%で,本剤300mg単回投与時には4時間の血液透析により投与量の約10%が除去された。
16.7 薬物相互作用
In vivoにおいて認められる濃度よりもはるかに高濃度(約300倍)において,テノホビルはヒトチトクロームP450分子種(CYP3A4,CYP2D6,CYP2C9又はCYP2E1)を阻害しなかったが,CYP1Aをわずかに(6%)阻害した。
本剤と主な薬剤との併用による,薬物動態への影響を下表に示す(表2及び表3)。
また,表4に本剤とジダノシンとの相互作用を示す。[10.2 参照]
| 併用薬 | 併用薬の用量 | 例数 | 他剤併用時/非併用時のテノホビルの 薬物動態パラメータ変化率 (%)(90%信頼区間) |
||
|---|---|---|---|---|---|
| Cmax | AUC | Cmin | |||
| アバカビル | 300mg 1回 |
8 | ⇔ | ⇔ | - |
| ラミブジン | 150mg 1日2回,7日間 |
15 | ⇔ | ⇔ | ⇔ |
| ジダノシン (腸溶剤) [10.2 参照] |
400mg 1回 |
25 | ⇔ | ⇔ | ⇔ |
| ジダノシン (制酸剤含有) [10.2 参照] |
250あるいは 400mg注3) 1日1回,7日間 |
14 | ⇔ | ⇔ | ⇔ |
| インジナビル | 800mg 1日3回,7日間 |
13 | ↑14 (↓3~↑33) |
⇔ | ⇔ |
| ロピナビル・リトナビル [10.2 参照] |
400/100mg 1日2回,14日間 |
24 | ⇔ | ↑32 (↑25~↑38) |
↑51 (↑37~↑66) |
| エファビレンツ | 600mg 1日1回,14日間 |
29 | ⇔ | ⇔ | ⇔ |
| アタザナビル [10.2 参照] |
400mg 1日1回,14日間 |
33 | ↑14 (↑8~↑20) |
↑24 (↑21~↑28) |
↑22 (↑15~↑30) |
| アデホビルピボキシル | 10mg 1回 |
22 | ⇔ | ⇔ | - |
| エムトリシタビン | 200mg 1日1回,7日間 |
17 | ⇔ | ⇔ | ⇔ |
| ネルフィナビル | 1,250mg 1日2回,14日間 |
29 | ⇔ | ⇔ | ⇔ |
| サキナビル+ リトナビル |
1,000/100mg 1日2回,14日間 |
35 | ⇔ | ⇔ | ↑23 (↑16~↑30) |
| ダルナビル+ リトナビル [10.2 参照] |
300/100mg 1日2回 |
12 | ↑24 (↑8~↑42) |
↑22 (↑10~↑35) |
↑37 (↑19~↑57) |
| レジパスビル・ ソホスブビル注4) [10.2 参照] |
90/400mg 1日1回,10日間 |
24 | ↑47 (↑37~↑58) |
↑35 (↑29~↑42) |
↑47 (↑38~↑57) |
| レジパスビル・ ソホスブビル注5) [10.2 参照] |
23 | ↑64 (↑54~↑74) |
↑50 (↑42~↑59) |
↑59 (↑49~↑70) |
|
| レジパスビル・ ソホスブビル注6) [10.2 参照] |
90/400mg 1日1回,14日間 |
15 | ↑79 (↑56~↑104) |
↑98 (↑77~↑123) |
↑163 (↑132~↑197) |
| レジパスビル・ ソホスブビル注7) [10.2 参照] |
90/400mg 1日1回,10日間 |
14 | ↑32 (↑25~↑39) |
↑40 (↑31~↑50) |
↑91 (↑74~↑110) |
| レジパスビル・ ソホスブビル注8) [10.2 参照] |
90/400mg 1日1回,10日間 |
29 | ↑61 (↑51~↑72) |
↑65 (↑59~↑71) |
↑115 (↑105~↑126) |
- 上昇:↑,低下:↓,不変:⇔,未算出:-
- 注3) 体重60kg未満:250mg,60kg以上:400mg
- 注4) アタザナビル硫酸塩,リトナビル,エムトリシタビン・テノホビル ジソプロキシルフマル酸塩配合錠との併用
- 注5) ダルナビル エタノール付加物,リトナビル,エムトリシタビン・テノホビル ジソプロキシルフマル酸塩配合錠との併用
- 注6) エファビレンツ・エムトリシタビン・テノホビル ジソプロキシルフマル酸塩配合錠との併用
- 注7) リルピビリン塩酸塩・テノホビル ジソプロキシルフマル酸塩・エムトリシタビン配合錠との併用
- 注8) ドルテグラビル+エムトリシタビン・テノホビル ジソプロキシルフマル酸塩を用いた薬物動態試験
| 併用薬 | 併用薬の用量 | 例数 | 他剤併用時/非併用時の併用薬の 薬物動態パラメータ変化率 (%)(90%信頼区間) |
||
|---|---|---|---|---|---|
| Cmax | AUC | Cmin | |||
| アバカビル | 300mg 1回 |
8 | ↑12 (↓1~↑26) |
⇔ | - |
| ラミブジン | 150mg 1日2回,7日間 |
15 | ↓24 (↓34~↓12) |
⇔ | ⇔ |
| 経口避妊薬 | エチニルエストラジオール・ノルゲスチメート 1日1回,7日間 |
20 | ⇔ | ⇔ | ⇔ |
| インジナビル | 800mg 1日3回,7日間 |
12 | ↓11 (↓30~↑12) |
⇔ | ⇔ |
| ロピナビル・ リトナビル [10.2 参照] |
400/100mg 1日2回,14日間 |
24 | ⇔ | ⇔ | ⇔ |
| エファビレンツ | 600mg 1日1回,14日間 |
30 | ⇔ | ⇔ | ⇔ |
| アタザナビル [10.2 参照] |
400mg 1日1回,14日間 |
34 | ↓21 (↓27~↓14) |
↓25 (↓30~↓19) |
↓40 (↓48~↓32) |
| アタザナビル+ リトナビル [10.2 参照] |
300/100mg 1日1回,42日間 |
10 | ↓28 (↓50~↑5) |
↓25注9) (↓42~↓3) |
↓23注9) (↓46~↑10) |
| リバビリン | 600mg 1回 |
22 | ⇔ | ⇔ | - |
| アデホビルピボキシル | 10mg 1回 |
22 | ⇔ | ⇔ | - |
| エムトリシタビン | 200mg 1日1回,7日間 |
17 | ⇔ | ⇔ | ↑20 (↑12~↑29) |
| ネルフィナビル M8代謝物 |
1,250mg 1日2回,14日間 |
29 | ⇔ ⇔ |
⇔ ⇔ |
⇔ ⇔ |
| サキナビル | 1,000/100mg 1日2回,14日間 |
32 | ↑22 (↑6~↑41) |
↑29 (↑12~↑48) |
↑47 (↑23~↑76) |
| リトナビル | ⇔ | ⇔ | ↑23 (↑3~↑46) |
||
| ダルナビル [10.2 参照] |
300/100mg 1日2回 |
12 | ↑16 (↓6~↑42) |
↑21 (↓5~↑54) |
↑24 (↓10~↑69) |
- 上昇:↑,低下:↓,不変:⇔,算出不能:-
- 注9) HIV感染症患者において,本剤にアタザナビル300mg及びリトナビル100mgを併用した場合,アタザナビルのAUC及びCminは,アタザナビル400mgを単独投与した場合と比較してそれぞれ2.3倍及び4倍上昇した。
| ジダノシンの用量 /投与方法注10) |
本剤の |
例数 | ジダノシン空腹時400mg投与時に対する 薬物動態パラメータ変化率 (%)(90%信頼区間) |
||
|---|---|---|---|---|---|
| Cmax | AUC | ||||
| 制酸剤含有製剤 400mg注11) 1日1回,7日間 |
空腹時 ジダノシン投与後 1時間 |
14 | ↑28 (↑11~↑48) |
↑44 (↑31~↑59) |
|
| 腸溶剤 | 空腹時 400mg,1回 |
食後 ジダノシン投与後 2時間 |
26 | ↑48 (↑25~↑76) |
↑48 (↑31~↑67) |
| 食後 400mg,1回 |
ジダノシンと同時投与 | 26 | ↑64 (↑41~↑89) |
↑60 (↑44~↑79) |
|
| 空腹時 250mg,1回 |
食後 ジダノシン投与後 2時間 |
28 | ↓11 (↓22~↑3) |
⇔ | |
| 空腹時 250mg,1回 |
ジダノシンと同時投与 | 28 | ⇔ | ↑14 (0~↑31) |
|
| 食後 250mg,1回 |
ジダノシンと同時投与 | 28 | ↓29 (↓39~↓18) |
↓11 (↓23~↑2) |
|
- 上昇:↑,低下:↓,不変:⇔
- 注10) 食後投与の食事は軽食(約373kcal,20%が脂肪由来)
- 注11) 体重60kg以下の症例4例含む(ジダノシンは250mg投与)
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第III相試験(907試験)
抗レトロウイルス薬による治療を経験した患者550例を対象とし,継続中の抗レトロウイルス薬による治療に本剤(300mg 1日1回投与)又はプラセボを併用した多施設二重盲検試験を実施した4)。患者の試験開始時の平均CD4リンパ球数は427cells/mm3,血漿中HIV-1 RNA量の中央値は2,340copies/mLであり,HIV-1感染症に対する前治療歴は平均5.4年であった。また,患者の平均年齢は42歳,85%が男性であり,69%が白人であった。
試験開始後48週までの血漿中HIV-1 RNA量の経時的変化(log10copies/mL)を図1に示す。なお,本試験では試験開始後24週目よりプラセボ投与群は,全て本剤投与へと変更された。
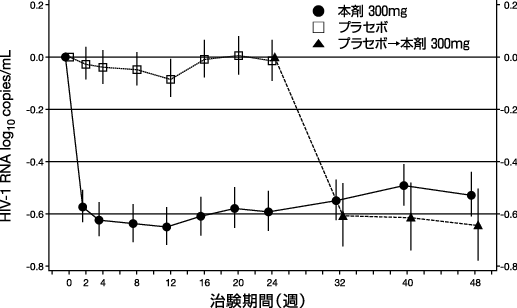
試験開始後24週及び48週の血漿中HIV-1 RNA量が<400copies/mLであった患者の比率は,本剤投与群で各々40%及び28%であり,プラセボ投与群では24週後で11%であった。さらに試験開始後24週の血漿中HIV-1 RNA量が<50copies/mLであった患者の比率は,本剤投与群で19%,プラセボ投与群で1%であった。
また,試験開始後24週のCD4リンパ球数の平均変化量は,本剤投与群及びプラセボ投与群で各々+11cells/mm3及び-5cells/mm3であり,本剤投与群の試験開始後48週の変化量は+4cells/mm3であった。
17.1.2 海外第III相試験(903試験)
抗レトロウイルス薬による治療を未経験の患者600例を対象とし,ラミブジン及びエファビレンツに本剤(300mg 1日1回投与)又はサニルブジンを併用した多施設二重盲検試験を実施した。患者の試験開始時の平均CD4リンパ球数は279cells/mm3,血漿中HIV-1 RNA量の中央値は77,600copies/mL,血漿中HIV-1 RNA量が>100,000copies/mLの患者は43%,CD4リンパ球数が<200cells/mm3の患者は39%であった。患者の平均年齢は36歳,74%が男性であり,64%が白人であった。試験開始後48週及び144週の結果を表1に示す。
| 結果 | 48週評価 | 144週評価 | ||
|---|---|---|---|---|
| 本剤 (299例) |
サニルブジン (301例) |
本剤 (299例) |
サニルブジン (301例) |
|
| 有効例注1) | 79% | 82% | 68% | 62% |
| 無効例注2) | 6% | 4% | 10% | 8% |
| 再上昇例 | 5% | 3% | 8% | 7% |
| 無反応例 | 0% | 1% | 0% | 0% |
| 他剤追加例 | 1% | 1% | 2% | 1% |
| 死亡例 | <1% | 1% | <1% | 2% |
| 有害事象による中止例 | 6% | 6% | 8% | 13% |
| その他の理由による中止例注3) | 8% | 7% | 14% | 15% |
- 注1) 血漿中HIV-1 RNA量が<400copies/mLに至り試験開始後48週及び144週まで維持していた症例
- 注2) 血漿中HIV-1 RNA量が<400copies/mLに至らなかった症例及び至った後に再上昇した症例
- 注3) 患者追跡不能例,患者申出による脱落例,服薬不良例,プロトコール不遵守例等
本試験における試験開始後144週のHIV-1 RNA量が<50copies/mLであった患者の比率は本剤投与群で62%,サニルブジン投与群で58%であった。また,CD4リンパ球数の平均増加量は,本剤投与群で263cells/mm3,サニルブジン投与群で283cells/mm3であった。
18. 薬効薬理
18.1 作用機序
テノホビル ジソプロキシルフマル酸塩は,アデノシン一リン酸の非環状ヌクレオシド・ホスホン酸ジエステル誘導体である。テノホビル ジソプロキシルフマル酸塩からテノホビルへの変換には,ジエステルの加水分解が必要であり,その後細胞内酵素によりリン酸化を受け,テノホビル二リン酸となる5)。テノホビル二リン酸は,HIV-1逆転写酵素の基質であるデオキシアデノシン5’-三リン酸と競合すること及びDNAに取り込まれた後にDNA鎖伸長を停止させることにより,HIV-1逆転写酵素の活性を阻害する。哺乳類のDNAポリメラーゼα,β及びミトコンドリアDNAポリメラーゼγに対するテノホビル二リン酸の阻害作用は弱い6)。
18.2 抗ウイルス作用(in vitro)
HIV-1の実験室株及び臨床分離株に対するテノホビルの抗ウイルス活性を,ヒトリンパ芽球様細胞株,単球/マクロファージ初代培養細胞及び末梢血リンパ球において評価した。テノホビルのIC50値は,0.04μM~8.5μMの範囲であった。
18.3 薬剤耐性
18.3.1 In vitro試験
テノホビルに対する感受性が低下したHIV-1分離株をin vitro試験により選択した結果,これらのウイルスは逆転写酵素遺伝子にK65R変異が発現しており,テノホビルに対する感受性が3~4倍低下していた。
18.3.2 臨床成績
本剤を他の抗レトロウイルス薬と併用した患者から,テノホビルに対して感受性が低下したHIV-1株が分離された。治療を経験した患者では,本剤による試験開始後96週までのウイルス学的失敗例304例のうち14例からテノホビル耐性株が認められた。分離された耐性株を遺伝子型解析したところ,HIV-1逆転写酵素遺伝子にK65R変異が発現していた。また,抗レトロウイルス薬による治療を未経験の患者に対する本剤+ラミブジン+エファビレンツの3剤併用療法では,試験開始後144週までのウイルス学的失敗例47例のうち,8例からテノホビル耐性株が確認された。
(1) 抗レトロウイルス療法経験例における抗ウイルス作用(遺伝子型解析)
治療経験を有する患者を対象として,本剤投与前におけるウイルス遺伝子型が本剤のウイルス学的効果に及ぼす影響を検討した(222例)。本剤投与開始前の患者から分離したHIV-1株の94%に1ヵ所以上のNRTI変異が検出され,また,評価した患者の大部分において,プロテアーゼ阻害薬又は非核酸系逆転写酵素阻害薬に関連した変異が認められた。特定の変異あるいは変異数とHIV-1 RNA量の変化との関係を表1に示した。
| 試験開始前のジドブジン |
HIV-1 RNA量の変化注2)(例数) | |
|---|---|---|
| 本剤300mg | プラセボ | |
| なし | -0.80(68) | -0.11(29) |
| あり | -0.50(154) | 0(81) |
| 1-2 | -0.66(55) | -0.04(33) |
| M41LあるいはL210Wを 含む3個以上の変異 |
-0.21(57) | +0.01(29) |
| M41LあるいはL210W以外の 3個以上の変異 |
-0.67(42) | +0.07(19) |
- 注1) 逆転写酵素のM41L,D67N,K70R,L210W,T215Y/F又はK219Q/E/N変異
- 注2) 試験開始時から24週までのHIV-1 RNA量時間加重平均の変化をlog10copies/mLで示した。
本剤投与により,M41L又はL210Wを含む3個以上のジドブジン関連変異を伴う場合にウイルス学的効果は低下したが,プラセボと比較した場合には効果が認められた。
一方,M184V(ラミブジン+エムトリシタビン+アバカビル関連変異)の変異は本剤のウイルス学的効果に影響を与えず,M184V変異があってもジドブジン関連変異が無ければ,プラセボ群と比較し0.84log10copies/mL減少した。また,K65Rの変異により本剤のウイルス学的効果が減少する傾向が認められた。
(2) 抗レトロウイルス療法経験例における抗ウイルス作用(表現型解析)
治療経験を有する患者を対象に,本剤投与前におけるウイルス表現型が本剤投与のウイルス学的効果に及ぼす影響を検討した(100例)。本剤投与開始前の患者から分離したHIV-1株の本剤に対する感受性と本剤のウイルス学的効果とには相関が見られ,その関係を表2に示した。
| 試験開始前の |
HIV-1 RNA量の変化注4)(例数) |
|---|---|
| ≦1 1<~≦3 3<~≦4 |
-0.74(35) -0.56(49) -0.3(7) |
| ≦4 >4 |
-0.61(91) -0.12(9) |
- 注3) 野生型からの感受性変化(倍数)
- 注4) 試験開始時から24週までのHIV-1 RNA量時間加重平均の変化をlog10copies/mLで示した。
(3) 抗レトロウイルス療法未経験例における薬剤耐性
903試験におけるウイルス学的失敗例から分離したHIV-1株では,エファビレンツ関連変異及びラミブジン関連変異が最も高頻度に認められ,本剤投与群とサニルブジン投与群との間に差は認められなかった。K65R変異は試験開始後144週までに本剤投与群の8例及びサニルブジン投与群の2例から分離したHIV-1株に認められたが,本剤投与群の8例のうち,7例では48週までに,1例では96週までに発現した。K65R以外にテノホビル耐性に関連する変異は認められなかった。
18.4 交差耐性
テノホビルで選択されるK65R変異は,アバカビル,ジダノシン及びザルシタビンにより治療された症例から分離したHIV-1株でも認められている。この変異株はエムトリシタビンやラミブジンに対する感受性も低下していたことから,K65R変異を持つウイルスを有する患者では,これらの薬剤間で交差耐性を起こす可能性がある。
ジドブジン関連変異(M41L,D67N,K70R,L210W,T215Y/F又はK219Q/E/N)を有するHIV-1分離株に対するテノホビルの活性をin vitroで評価した。20例から分離した複数(平均3ヵ所)のジドブジン関連変異を有するHIV-1臨床分離株において,テノホビルに対する感受性は3.1倍低下していた。また,T69S変異の後に二アミノ酸が挿入される変異を持つ多剤耐性株においても,テノホビルに対する感受性は低下していた7)。
19. 有効成分に関する理化学的知見
| 一般的名称 | テノホビル ジソプロキシルフマル酸塩(Tenofovir Disoproxil Fumarate) |
|---|---|
| 化学名 | Bis(isopropoxycarbonyloxymethyl){[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}phosphonate monofumarate |
| 分子式 | C19H30N5O10P・C4H4O4 |
| 分子量 | 635.51 |
| 性状 | 白色~帯黄白色の結晶性の粉末であり,メタノール,エタノール(95)にやや溶けやすく,アセトン,水にやや溶けにくく,ジエチルエーテルにほとんど溶けない。 |
| 化学構造式 | 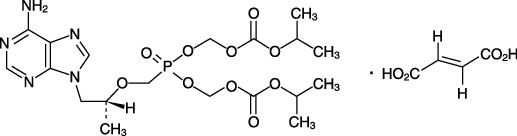 |
| 融点 | 114~118℃ |
| 分配係数 | 1.25(1-オクタノール/pH6.5のリン酸塩緩衝液) |
20. 取扱い上の注意
開栓後は,湿気を避けて保存すること。
22. 包装
30錠/瓶[乾燥剤入り]
23. 主要文献
- 社内資料:1278-005試験(VAD承認年月日:2004.3.25) [VAD-001]
- Benaboud S. et al. Antimicrob. Agents Chemother. 2011;55(3):1315-1317. [VAD-002]
- 中道昇,他. 新薬と臨牀. 2005;54(8):941-948. [VAD-003]
- Squires K. et al. Ann. Intern. Med. 2003;139(5):313-320. [VAD-004]
- Robbins B. L. et al. Antimicrob. Agents Chemother. 1998;42(3):612-617. [VAD-005]
- Cihlar T. et al. Antivir. Chem. Chemother. 1997;8(3):187-195. [VAD-006]
- Miller M. D. et al. Nucleosides Nucleotides Nucleic Acids. 2001;20(4-7):1025-1028. [VAD-007]
24. 文献請求先及び問い合わせ先
ギリアド・サイエンシズ株式会社
メディカルサポートセンター
〒100-6616 東京都千代田区丸の内一丁目9番2号
グラントウキョウサウスタワー
フリーダイアル 0120-506-295
FAX 03-5958-2959
受付時間:9:00~17:30(土・日・祝日及び会社休日を除く)
26. 製造販売業者等
26.1 製造販売元
ギリアド・サイエンシズ株式会社
東京都千代田区丸の内1-9-2
グラントウキョウサウスタワー