エジュラント錠25mgの添付文書
- 商品名:
- エジュラント錠25mg
- 一般名:
- リルピビリン
- 略称 :
- RPV

添付文書の読み方
ここで提供している添付文書情報は、2025年2月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
抗ウイルス化学療法剤[非ヌクレオシド系逆転写酵素阻害剤(NNRTI)]
リルピビリン
- **2024年9月改訂(第6版、再審査結果)
- *2023年8月改訂
- 劇薬
- 処方箋医薬品※
※注意-医師等の処方箋により使用すること
| 日本標準商品分類番号 | 87625 |
|---|
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 36ヵ月 |
| 承認番号 | 22400AMX00687000 |
|---|---|
| 販売開始 | 2012年6月 |
2. 禁忌(次の患者には投与しないこと)
2.1
本剤の成分に対し過敏症の既往歴のある患者
**2.2
リファンピシン、カルバマゼピン、フェノバルビタール、フェニトイン、フェニトイン・フェノバルビタール、ホスフェニトイン、デキサメタゾン(全身投与)(単回投与を除く)、セイヨウオトギリソウ(St.John’s Wort、セント・ジョーンズ・ワート)含有食品、プロトンポンプ阻害剤(オメプラゾール、ランソプラゾール、アスピリン・ランソプラゾール、ラベプラゾール、エソメプラゾール、ボノプラザンフマル酸塩、アスピリン・ボノプラザンフマル酸塩)を投与中の患者[10.1 参照]
3. 組成・性状
3.1 組成
| 有効成分 | (1錠中) リルピビリン塩酸塩27.5mg(リルピビリンとして25mg)含有 |
|---|---|
| **添加剤 | 乳糖水和物、クロスカルメロースナトリウム、ポビドン、ポリソルベート20、ケイ酸処理結晶セルロース、ステアリン酸マグネシウム、マクロゴール4000、ヒプロメロース、トリアセチン、酸化チタン |
3.2 製剤の性状
| 外形 | 表面 | 裏面 | 側面 |
|---|---|---|---|
 |
 |
 |
|
| 大きさ | 直径(mm) | 厚さ(mm) | 重量(g) |
| 6.4 | 3.4 | 0.1 | |
| 識別コード | TMC 25 | ||
| 色・剤形 | 白色~オフホワイトのフィルムコーティング錠 | ||
4. 効能又は効果
HIV-1感染症
5. 効能又は効果に関連する注意
〈併用薬共通〉
5.1
本剤による治療にあたっては、患者の治療歴及び可能な場合には薬剤耐性検査(遺伝子型解析あるいは表現型解析)を参考にすること。
〈カボテグラビル経口剤以外の抗HIV薬併用時〉
5.2
抗HIV薬の治療経験がなく、HIV-1 RNA量100,000 copies/mL以下の患者に使用すること。[17.1.1 参照],[17.1.2 参照]
5.3
海外臨床第III相試験の併合解析において本剤によるウイルス学的失敗例では、エファビレンツによるウイルス学的失敗例よりも、ラミブジン/エムトリシタビンへの耐性変異の発現割合が高かった。また、ベースラインCD4陽性リンパ球数が少ない被験者(<200cells/μL)では、ベースラインCD4陽性リンパ球数が多い被験者(≧200cells/μL)と比べてウイルス学的失敗例の割合が高かった。本剤による治療開始時には、これらの情報について考慮すること。[17.参照]
〈カボテグラビル経口剤併用時〉
5.4
本剤は、ウイルス学的失敗の経験がなく、切り替え前6ヵ月間以上においてウイルス学的抑制(ヒト免疫不全ウイルス[HIV]-1 RNA量が50copies/mL未満)が得られており、リルピビリン及びカボテグラビルに対する耐性関連変異を持たず、本剤への切り替えが適切であると判断される抗HIV薬既治療患者に使用すること。[17.1.3 参照],[17.1.4 参照],[17.1.5 参照]
5.5
本剤は以下の場合に使用すること。
長期作用型の薬剤であるリルピビリン注射剤の投与に先立って、経口導入としてリルピビリンへの忍容性を確認する。
リルピビリン注射剤を予定するスケジュール通りに投与できない場合の代替薬として使用する。
6. 用法及び用量
通常、成人にはリルピビリンとして1回25mgを1日1回食事中又は食直後に経口投与する。投与に際しては、必ず他の抗HIV薬と併用すること。
7. 用法及び用量に関連する注意
〈併用薬共通〉
7.1
本剤とリファブチンを併用する場合は、本剤を50mg 1日1回に増量すること。なお、リファブチンの併用を中止した場合は、本剤を25mg 1日1回に減量すること。[10.2 参照],[16.7 参照]
〈カボテグラビル経口剤以外の抗HIV薬併用時〉
7.2
非ヌクレオシド系逆転写酵素阻害剤(NNRTI)を2剤併用したときの有用性が示されていない。他のNNRTIとの併用は避けることが望ましい。
〈カボテグラビル経口剤併用時〉
7.3
リルピビリン注射剤及びカボテグラビル注射剤の併用療法の経口導入として用いる場合には、本剤をカボテグラビル経口剤との併用により1ヵ月間(少なくとも28日間)を目安に経口投与し、リルピビリン及びカボテグラビルに対する忍容性を確認すること。
7.4
リルピビリン注射剤を投与予定日の7日後までに投与できない場合は、本剤による代替投与が可能であるが、以下の点に留意すること。
代替投与可能な期間は2ヵ月間までであること。本剤による代替期間が2ヵ月間を超える場合は、他の抗HIV薬へ切り替えることを考慮すること。
リルピビリン注射剤を再開する際にはリルピビリン注射剤の電子添文を参照すること。
8. 重要な基本的注意
*8.1
本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又は患者に代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。
8.1.1
本剤はHIV感染症の根治療法薬ではないことから、日和見感染を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。
8.1.2
本剤の長期投与による影響については、現在のところ不明であること。
8.1.3
本剤を処方どおりに毎日服用すること。また、担当医の指示なしに用量を変更したり、服用を中止したりしないこと。
8.1.4
本剤は併用薬剤と相互作用を起こすことがあるため、服用中のすべての薬剤を担当医に報告すること。また、本剤で治療中に新たに他の薬剤を服用する場合、事前に担当医に相談すること。
8.2
本剤を含む抗HIV薬の多剤併用療法を行った患者で、免疫再構築症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 不整脈を起こしやすい患者
低カリウム血症、著しい徐脈、急性心筋虚血、うっ血性心不全、先天性QT延長症候群等の患者では、QT延長により不整脈が発現するおそれがある。本剤75mg及び300mg投与時にQT延長が認められている。[10.2 参照],[17.3.1 参照]
9.1.2 B型及び/又はC型肝炎ウイルス重複感染患者
定期的な肝機能検査を行うなど、観察を十分に行い、異常が認められた場合には、投与を中止するなど適切な処置を行うこと。海外第III相試験において、これらの患者では、肝臓関連有害事象(臨床検査値異常を含む)の発現頻度が非重複感染患者より高かった[重複感染患者33.3%(18/54例)、非重複感染患者4.9%(31/632例)]。
9.5 妊婦
9.5.1
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
9.5.2
妊娠中期及び妊娠後期の妊婦に本剤を投与したとき、出産後と比較し、リルピビリンの血中濃度低下が認められている。[16.6.4 参照]
9.6 授乳婦
授乳を避けさせること。リルピビリンは、動物実験(ラット)で乳汁中へ移行することが報告されているが、ヒトにおける乳汁への移行は不明である。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。本剤は主として肝臓で代謝されるが、一般に肝機能が低下していることが多いため、高い血中濃度が持続するおそれがある。
10. 相互作用
本剤は主にCYP3Aにより代謝される。
**10.1 併用禁忌(併用しないこと)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| リファンピシン1) リファジン [2.2 参照],[16.7 参照] |
本剤の血中濃度が低下し、本剤の効果が減弱するおそれがある。 | これらの薬剤のCYP3A誘導作用により、本剤の代謝が促進される。 |
|
本剤の血中濃度が低下し、本剤の効果が減弱するおそれがある。 | |
| デキサメタゾン(全身投与)(単回投与を除く) デカドロン等 [2.2 参照] |
||
| セイヨウオトギリソウ(St.John’s Wort、セント・ジョーンズ・ワート)含有食品 [2.2 参照] |
||
プロトンポンプ阻害剤
|
本剤の血中濃度が低下し、本剤の効果が減弱するおそれがある。 | 胃内のpH上昇により、本剤の吸収が低下する。 |
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| リファブチン3) [7.1 参照],[16.7 参照] |
本剤の血中濃度が低下し、本剤の効果が減弱するおそれがある。 | リファブチンのCYP3A誘導作用により、本剤の代謝が促進される。 |
| H2遮断剤 ファモチジン4) シメチジン ニザチジン ラニチジン [16.7 参照] |
本剤の血中濃度が低下し、本剤の効果が減弱するおそれがある。これらの薬剤は、本剤投与の12時間以上前又は4時間以上後に投与すること。 | 胃内のpH上昇により、本剤の吸収が低下する。 |
| 制酸剤 乾燥水酸化アルミニウムゲル 沈降炭酸カルシウム等 |
本剤の血中濃度が低下し、本剤の効果が減弱するおそれがある。これらの薬剤は、本剤投与の2時間以上前又は4時間以上後に投与すること。 | 胃内のpH上昇により、本剤の吸収が低下する。 |
| クラリスロマイシン エリスロマイシン |
本剤の血中濃度が上昇する可能性がある。代替としてアジスロマイシン等を考慮すること。 | これらの薬剤のCYP3A阻害作用により、本剤の代謝が阻害される。 |
| メサドン5) [16.7 参照] |
メサドンの血中濃度が低下することがある。 | 機序不明 |
| QT延長を起こすことが知られている薬剤 アミオダロン ソタロール等 [9.1.1 参照],[17.3.1 参照] |
QT延長、心室性頻拍(Torsade de Pointesを含む)が発現するおそれがある。 | 本剤75mg及び300mg投与時にQT延長が認められている。 |
| ヌクレオシド/ヌクレオチド系逆転写酵素阻害剤(NRTI/NtRTI) | ||
| ジダノシン6) | 本剤(食直後投与)とジダノシン400mg 1日1回(空腹時投与)を併用したとき、本剤及びジダノシンの薬物動態に影響はみられなかった。本剤とジダノシンを併用するときは用量を調節する必要はないが、ジダノシンは空腹時に服用することが望ましいため、本剤服用(食事中又は食直後)の1時間前又は2時間後にジダノシンを投与するなど本剤と同時に投与しないこと。 | |
| テノホビル7) | テノホビル(フマル酸テノホビルジソプロキシル300mg 1日1回)を併用したとき、テノホビルのCmax及びAUCがそれぞれ19%及び23%増加した。本剤とテノホビルを併用するとき、用量を調節する必要はない。 | 機序不明 |
| プロテアーゼ阻害剤(PI) | ||
| ダルナビル/リトナビル8) | ダルナビル/リトナビル800/100mgを1日1回併用したとき、本剤のCmax及びAUCがそれぞれ79%及び130%増加した。ダルナビル/リトナビルと併用する場合には、用量を調節する必要はない。 | ダルナビル/リトナビルのCYP3A阻害作用により、本剤の代謝が阻害される。 |
| ロピナビル・リトナビル配合剤9) | ロピナビル・リトナビル400・100mgを1日2回併用したとき、本剤のCmax及びAUCがそれぞれ29%及び52%増加した。ロピナビル・リトナビルと併用する場合には、用量を調節する必要はない。 | ロピナビル・リトナビルのCYP3A阻害作用により、本剤の代謝が阻害される。 |
| 他のPI アタザナビル/リトナビル ホスアンプレナビル/リトナビル |
PIとの併用により、本剤の血中濃度が上昇する可能性がある。また、本剤は、PIの血中濃度に影響を与えないと推察される。 | PIのCYP3A阻害作用により、本剤の代謝が阻害される。 |
| インテグラーゼ阻害剤 | ||
| ラルテグラビル10) | ラルテグラビル400mgを1日2回併用したとき、本剤の薬物動態に影響を与えなかった。ラルテグラビルのCmax及びAUCがそれぞれ10%及び9%増加した。ラルテグラビルと併用する場合には、用量を調節する必要はない。 | 機序不明 |
11. 副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.2 その他の副作用
〈カボテグラビル経口剤以外の抗HIV薬併用時〉
| 5%以上 | 5%未満 | 頻度不明 | |
|---|---|---|---|
| 免疫系障害 | 免疫再構築症候群 | ||
| 代謝及び栄養障害 | 食欲減退 | 体脂肪の再分布/蓄積 | |
| 精神障害 | 不眠症、異常な夢、うつ病 | 睡眠障害、抑うつ気分 | |
| 神経系障害 | 頭痛、浮動性めまい | 傾眠 | |
| 胃腸障害 | 悪心、腹痛、嘔吐 | 腹部不快感、口内乾燥 | |
| 皮膚及び皮下組織障害 | 発疹 | ||
| 一般・全身障害及び投与部位の状態 | 疲労 | ||
| 臨床検査 | 低リン酸血症、低ナトリウム血症、高ナトリウム血症、白血球数減少、AST増加、ALT増加、高ビリルビン血症、総コレステロール増加、低血糖、高血糖、LDLコレステロール増加、膵型アミラーゼ増加、リパーゼ増加 | ALP増加、ヘモグロビン減少、トリグリセリド増加 |
〈カボテグラビル経口剤併用時〉
| 1~10%未満 | 1%未満 | 頻度不明 | |
|---|---|---|---|
| 精神・神経系 | 頭痛、不安、異常な夢、不眠症、浮動性めまい | うつ病、傾眠 | |
| 消化器 | 悪心、下痢 | 嘔吐、腹痛、鼓腸 | |
| 皮膚 | 発疹 | ||
| 筋骨格 | 筋肉痛 | ||
| 全身症状 | 発熱、疲労、無力症、倦怠感 | ||
| 肝臓 | 肝機能障害 | ||
| 臨床検査 | 体重増加、トランスアミナーゼ上昇、リパーゼ増加 | 総ビリルビン上昇 |
13. 過量投与
13.1 処置
本剤は透析により除去されない。
16. 薬物動態
16.1 血中濃度
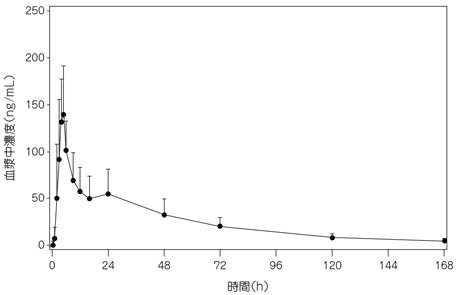
16.1.1 日本人における成績
健康成人に本剤25mgを食後に単回経口投与したとき、血漿中リルピビリン濃度は投与後5時間(中央値)に最高血漿中濃度[144.3ng/mL(平均値)]に達し、約43時間(平均値)の消失半減期で消失した。
平均AUC∞は4542ng・h/mLであった(表1、図1)。11)
| 薬物動態パラメータ | N=8 |
|---|---|
| Cmax(ng/mL) | 144.3(49.66) |
| tmax(h) | 5.00[2.00~6.00] |
| AUC∞(ng・h/mL) | 4542(2001) |
| t1/2(h) | 43.0(10.9) |
平均値(標準偏差),tmax:中央値[範囲]
16.1.2 外国人における成績
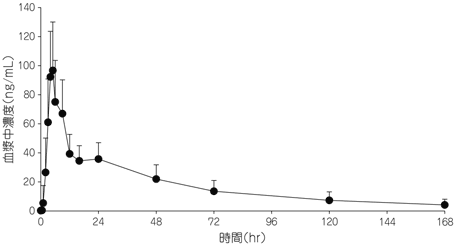
健康成人に本剤25mgを食後に単回経口投与したとき、血漿中リルピビリン濃度は投与後4~5時間(中央値)に最高血漿中濃度[109ng/mL(平均値)]に達し、約45時間(平均値)の消失半減期で消失した。平均AUC∞は3403ng・hr/mLであった(図2)。12)
抗HIV薬による治療経験のないHIV-1感染患者に、本剤25mgを1日1回反復経口投与した第III相試験の成績を用いた母集団薬物動態解析より得た血漿中リルピビリンの薬物動態パラメータ(推定値)を表2に示す。HIV-1感染患者における血漿中リルピビリンの曝露量は健康成人より低値であった。12)
| 薬物動態パラメータ | N=679 | |
|---|---|---|
| AUC24 (ng・hr/mL) |
平均値(標準偏差) 中央値[範囲] |
2235(851) 2096[198~7307] |
| C0 (ng/mL) |
平均値(標準偏差) 中央値[範囲] |
78(35) 73[2~288] |
16.2 吸収
16.2.1 食事の影響
健康成人に本剤75mgを、空腹時に単回経口投与したときの血漿中リルピビリンのAUCは、食直後に単回経口投与したときと比較して約40%低かった。また、高蛋白質栄養飲料摂取後に本剤75mgを経口投与したときの血漿中リルピビリンのAUCは、食直後(標準食)に経口投与したときと比較して50%低かった。13)(外国人データ)
16.3 分布
In vitro試験におけるリルピビリンの血漿蛋白結合率は約99.7%であり、主にアルブミンに結合した(平衡透析法)。14)
16.4 代謝
In vitro試験で、リルピビリンは主にCYP3Aにより代謝された。15)
16.5 排泄
健康成人に14C-リルピビリン(液剤)150mgを単回経口投与したとき、投与した総放射能の85%(平均値)が糞中、6.1%(平均値)が尿中から回収された。糞中及び尿中の未変化体の割合は、それぞれ投与量の25%(平均値)及び1%未満であった。16) (外国人データ)
16.6 特定の背景を有する患者
16.6.1 肝機能障害患者
軽度肝機能障害(Child-PughスコアA、8例)及び中等度肝機能障害(Child-PughスコアB、8例)患者に本剤25mgを1日1回反復経口投与したときの血漿中リルピビリンのAUC24は、健康成人と比較してそれぞれ47%及び5%高かった(表3)。軽度肝機能障害及び中等度肝機能障害患者に本剤を投与するとき、用量を調節する必要はない。なお、重度肝機能障害患者(Child-PughスコアC)を対象とした試験は実施していない。17) (外国人データ)
| 薬物動態パラメータ | 健康成人 | 肝機能障害患者 | 最小二乗平均の比 [90%信頼区間] |
|---|---|---|---|
| 軽度肝機能障害患者 | |||
| n | 8 | 8 | - |
| Cmax ng/mL | 144.3(35.70) | 187.0(66.31) | 1.268[0.9804~1.641] |
| tmax hr | 5.0[3.0~12.0] | 5.0[2.0~24.0] | - |
| AUC24 ng∙hr/mL | 2152(538.1) | 3206(1080) | 1.467[1.144~1.881] |
| t1/2 hr | 60.59(20.03) | 80.82(33.17)a) | - |
| 中等度肝機能障害患者 | |||
| n | 8 | 8 | - |
| Cmax ng/mL | 146.8(30.21) | 143.5(49.69) | 0.9496[0.7514~1.200] |
| tmax hr | 5.0[3.0~5.0] | 20.0[2.0~24.0] | - |
| AUC24 ng∙hr/mL | 2318(385.9) | 2525(851.2) | 1.052[0.8379~1.320] |
| t1/2 hr | 56.01(21.31) | 90.56(37.04)b) | - |
a): n=7, b): n=5
平均値(標準偏差),tmax:中央値[範囲]
16.6.2 B型肝炎ウイルス及び/又はC型肝炎ウイルス重複感染患者
母集団薬物動態解析の結果、B型肝炎ウイルス及び/又はC型肝炎ウイルスとHIV-1の重複感染患者の血漿中リルピビリンのAUC24及びC0に、臨床上問題となる影響はなかった。(外国人データ)
16.6.3 腎機能障害患者
腎機能障害患者を対象とした試験は実施していないが、リルピビリンの腎排泄は限定的であるため、腎機能障害によりリルピビリンの排泄にほとんど影響を及ぼさないと推察される。リルピビリンは血漿蛋白結合率が高いことから、血液透析や腹膜透析により除去される可能性は低い。14),16) )(外国人データ)
16.6.4 妊婦、産婦への投与
妊娠中期のHIV-1感染患者(15例)に、本剤25mgを1日1回投与したとき、リルピビリンのCmax、AUC24h及びCminは、出産後(6~12週;11例)と比較してそれぞれ21%、29%及び35%減少し、妊娠後期(13例)では、それぞれ20%、31%及び42%減少した。(外国人データ)[9.5.2 参照]
16.7 薬物相互作用
本剤と主な薬剤の併用による薬物動態への影響を下表に示す(表4及び表5)。(外国人データ)[7.1 参照],[10. 参照]
なお、アバカビル、エムトリシタビン、ラミブジン、サニルブジン、ジドブジンは主に腎排泄型であり、本剤と排泄経路が異なる。本剤はこれらの薬剤と相互作用を示さないと推察される。
| 併用薬 | 併用薬の用量 | 例数 | リルピビリンの薬物動態パラメータの比 併用時/単独投与時(%)(90%信頼区間) |
||
|---|---|---|---|---|---|
| Cmax | AUC | Cmin | |||
| ジダノシン6) | 400mg 1日1回 |
14~21 | 100 (90-110) |
100 (95-106) |
100 (92-109) |
| テノホビル7) | 300mg 1日1回 |
15~16 | 96 (81-113) |
101 (87-118) |
99 (83-116) |
| ダルナビル/リトナビル8) | 800mg/100mg 1日1回 |
14 | 179 (156-206) |
230 (198-267) |
278 (239-324) |
| ロピナビル・リトナビル配合剤9) | 400・100mg 1日2回 |
15 | 129 (118-140) |
152 (136-170) |
174 (146-208) |
| ラルテグラビルa)10) | 400mg 1日2回 |
23 | 112 (104-120) |
112 (105-119) |
103 (96-112) |
| リファブチン18) | 300mg 1日1回 |
14~16 | 65 (58-74) |
54 (50-58) |
51 (48-54) |
| リファブチンa)3) | 300mg 1日1回 |
10~18 | 69 (62-76) |
58 (52-65) |
52 (46-59) |
| リファブチンb)3) | 300mg 1日1回 |
17~18 | 143 (130-156)c) |
116 (106-126)c) |
93 (85-101)c) |
| ファモチジン4) | リルピビリン製剤 投与12時間前 40mg 1回 |
23~24 | 99 (84-116) |
91 (78-107) |
- |
| ファモチジン4) | リルピビリン製剤 投与2時間前 40mg 1回 |
22~23 | 15 (12-19) |
24 (20-28) |
- |
| ファモチジン4) | リルピビリン製剤 投与4時間後 40mg 1回 |
23~24 | 121 (106-139) |
113 (101-127) |
- |
| リファンピシン1) | 600mg 1日1回 |
15~16 | 31 (27-36) |
20 (18-23) |
11 (10-13) |
| ケトコナゾール19) | 400mg 1日1回 |
14~15 | 130 (113-148) |
149 (131-170) |
176 (157-197) |
| オメプラゾール2) | 20mg 1日1回 |
15~16 | 60 (48-73) |
60 (51-71) |
67 (58-78) |
| アセトアミノフェン20) | 500mg 1回 |
16 | 109 (101-118) |
116 (110-122) |
126 (116-138) |
| アトルバスタチン21) | 40mg 1日1回 |
16 | 91 (79-106) |
90 (81-99) |
90 (84-96) |
| クロルゾキサゾン22) | 500mg 1回 |
16 | 117 (108-127) |
125 (116-135) |
118 (109-128) |
| シルデナフィルd)23) | 50mg 1回 |
16 | 92 (85-99) |
98 (92-105) |
104 (98-109) |
| シメプレビルa) | 150mg 1日1回 |
21~23 | 104 (95-113) |
112 (105-119) |
125 (116-135) |
| カボテグラビルa)24) | 30mg 1日1回 |
11 | 96 (85-109) |
99 (89-109) |
92 (79-107) |
未算出:-
a)リルピビリン製剤25mg 1日1回投与時
b)リルピビリン製剤50mg 1日1回投与時
c)リルピビリン製剤25mgを単剤として投与したときとの比較
d)リルピビリン製剤75mg 1日1回投与時
| 併用薬 | 併用薬の用量 | 例数 | 併用薬の薬物動態パラメータの比 併用時/単独投与時(%)(90%信頼区間) |
||
|---|---|---|---|---|---|
| Cmax | AUC | Cmin | |||
| ジダノシン6) | 400mg 1日1回 |
13 | 96 (80-114) |
112 (99-127) |
- |
| テノホビル7) | 300mg 1日1回 |
15~16 | 119 (106-134) |
123 (116-131) |
124 (110-138) |
| ダルナビル8) | ダルナビル/リトナビル 800mg/100mg 1日1回 |
14~15 | 90 (81-100) |
89 (81-99) |
89 (68-116) |
| ロピナビル9) | ロピナビル・ リトナビル配合剤 400・100mg 1日2回 |
15 | 96 (88-105) |
99 (89-110) |
89 (73-108) |
| ラルテグラビルb)10) | 400mg 1日2回 |
23 | 110 (77-158) |
109 (81-147) |
127 (101-160) |
| リファブチン18) | 300mg 1日1回 |
16~17 | 103 (93-114) |
103 (97-109) |
101 (94-109) |
| リファンピシン1) | 600mg 1日1回 |
15~16 | 102 (93-112) |
99 (92-107) |
- |
| ケトコナゾール19) | 400mg 1日1回 |
14 | 85 (80-90) |
76 (70-82) |
34 (25-46) |
| オメプラゾール2) | 20mg 1日1回 |
15 | 86 (68-109) |
86 (76-97) |
- |
| アセトアミノフェン20) | 500mg 1回 |
16 | 97 (86-110) |
92 (85-99) |
- |
| エチニルエストラジオールb)25) | エチニルエストラジオール・ ノルエチステロン配合剤 0.035・1mg 1日1回 |
14~17 | 117 (106-130) |
114 (110-119) |
109 (103-116) |
| ノルエチステロンb)25) | エチニルエストラジオール・ ノルエチステロン配合剤 0.035・1mg 1日1回 |
14~17 | 94 (83-106) |
89 (84-94) |
99 (90-108) |
| アトルバスタチン21) | 40mg 1日1回 |
16 | 135 (108-168) |
104 (97-112) |
85 (69-103) |
| クロルゾキサゾン22) | 500mg 1回 |
16 | 98 (85-113) |
103 (95-113) |
- |
| シルデナフィルa)23) | 50mg 1回 |
15~16 | 93 (80-108) |
97 (87-108) |
- |
| R(-)メサドンb)5) | メサドン 60~100mg 1日1回 |
12~13 | 86 (78-95) |
84 (74-95) |
78 (67-91) |
| S(+)メサドンb)5) | メサドン 60~100mg 1日1回 |
12~13 | 87 (78-97) |
84 (74-96) |
79 (67-92) |
| メトホルミンb)26) | 850mg 1回 |
20 | 102 (95-110) |
99 (94-104) |
- |
| シメプレビルb) | 150mg 1日1回 |
20~21 | 110 (97-126) |
106 (94-119) |
96 (83-111) |
| ジゴキシンb)27) | 0.5mg 1回 |
21~22 | 106 (97-117) |
98 (93-104) |
- |
| カボテグラビルb)24) | 30mg 1日1回 |
11 | 105 (96-115) |
112 (105-119) |
114 (104-124) |
算出不能:-
a)リルピビリン製剤75mg 1日1回投与時
b)リルピビリン製剤25mg 1日1回投与時
17. 臨床成績
17.1 有効性及び安全性に関する試験
〈カボテグラビル経口剤以外の抗HIV薬併用時〉
17.1.1 抗HIV薬の使用経験のないHIV-1感染患者を対象とした海外臨床試験(第III相試験):C209試験(ECHO試験)及びC215試験(THRIVE試験)
抗HIV薬の使用経験のないHIV-1感染患者1368例を対象とし、リルピビリン(RPV)25mg及び背景治療(BR)の1日1回投与と、エファビレンツ(EFV)600mg及びBRの1日1回投与の無作為割付け、二重盲検の実薬対照による臨床第III相比較試験を2試験実施した。両試験はBRを除き同一のデザインであり、ECHO試験のBRはTDF/FTCとし、THRIVE試験ではABC/3TC、AZT/3TC、TDF/FTCから医師がBRを選択した。両試験の併合解析において両群の患者背景及び疾患特性に偏りはみられず、RPV+BR群686例の年齢中央値は36歳(範囲18-78)、男性が76%、人種は白人が61%、黒人又はアフリカ系アメリカ人24%、アジア人11%、その他が2%、規制により聴取不可が1%であった。ベースラインのHIV RNA量中央値は5.0 log10copies/mL(範囲2-7)、CD4陽性リンパ球数の中央値は249cells/μL(範囲1-888)、BRはTDF/FTCが80.2%、AZT/3TCが14.7%、ABC/3TCが5.1%であった。48週及び96週時の臨床成績を表1及び2に示す。33)[5.2 参照],[5.3 参照]
| 例数(%) | ECHO試験 | THRIVE試験 | ECHO試験及び THRIVE試験の併合解析 |
|||
|---|---|---|---|---|---|---|
| RPV+BR群 N=346 |
EFV+BR群 N=344 |
RPV+BR群 N=340 |
EFV+BR群 N=338 |
RPV+BR群 N=686 |
EFV+BR群 N=682 |
|
| 48週時 | ||||||
| ウイルス学的効果注1) HIV RNA量 <50copies/mL |
285 (82.4) |
286 (83.1) |
290 (85.3) |
274 (81.1) |
575 (83.8) |
560 (82.1) |
| ウイルス学的失敗注2) | 38 (11.0) |
15 (4.4) |
24 (7.1) |
18 (5.3) |
62 (9.0) |
33 (4.8) |
| 死亡 | 0 | 0 | 1 (0.3) |
3 (0.9) |
1 (0.1) |
3 (0.4) |
| 有害事象による 投与中止 |
6 (1.7) |
25 (7.3) |
8 (2.4) |
21 (6.2) |
14 (2.0) |
46 (6.7) |
| 他の理由による 投与中止 |
15 (4.3) |
19 (5.5) |
16 (4.7) |
20 (5.9) |
31 (4.5) |
39 (5.7) |
| 96週時 | ||||||
| ウイルス学的効果注1) HIV RNA量 <50copies/mL |
263 (76.0) |
271 (78.8) |
269 (79.1) |
258 (76.3) |
532 (77.6) |
529 (77.6) |
| ウイルス学的失敗注2) | 45 (13.0) |
16 (4.7) |
34 (10.0) |
24 (7.1) |
79 (11.5) |
40 (5.9) |
| 死亡 | 0 | 3 (0.9) |
1 (0.3) |
3 (0.9) |
1 (0.1) |
6 (0.9) |
| 有害事象による 投与中止 |
10 (2.9) |
29 (8.4) |
16 (4.7) |
23 (6.8) |
26 (3.8) |
52 (7.6) |
| 他の理由による 投与中止 |
28 (8.1) |
25 (7.3) |
20 (5.9) |
30 (8.9) |
48 (7.0) |
55 (8.1) |
注1)ITT-TLOVR:HIV RNA量<50copies/mLが連続して認められ48週又は96週時まで持続
注2)ウイルス学的再燃例(2回連続でHIV-1 RNA量<50copies/mLが認められ、その後48週又は96週までに2回連続でHIV-1 RNA量≧50copies/mLが認められた患者)又はウイルス学的非抑制例(48週又は96週までに2回連続したHIV-1 RNA量<50copies/mLが認められなかった患者)を含む。
| 48週時 | 96週時 | |||
|---|---|---|---|---|
| RPV+BR群 N=686 |
EFV+BR群 N=682 |
RPV+BR群 N=686 |
EFV+BR群 N=682 |
|
| ベースラインHIV RNA量(copies/mL)別ウイルス学的効果 | ||||
| ≦100,000 | 90.2% (332/368例) |
83.6% (276/330例) |
84.0% (309/368例) |
79.9% (263/329例) |
| >100,000 | 77.4% (246/318例) |
81.0% (285/352例) |
70.1% (223/318例) |
75.4% (266/353例) |
| ベースラインHIV RNA量(copies/mL)別ウイルス学的失敗 | ||||
| ≦100,000 | 4.3% (16/368例) |
3.3% (11/329例) |
5.7% (21/368例) |
3.6% (12/329例) |
| >100,000 | 15.4% (49/318例) |
6.8% (24/352例) |
18.2% (58/318例) |
7.9% (28/353例) |
| ベースラインCD4陽性リンパ球数(cells/μL)別ウイルス学的効果 | ||||
| <50 | 58.8% (20/34例) |
80.6% (29/36例) |
55.9% (19/34例) |
69.4% (25/36例) |
| ≧50、<200 | 80.4% (156/194例) |
81.7% (143/175例) |
71.1% (138/194例) |
74.9% (131/175例) |
| ≧200、<350 | 86.9% (272/313例) |
82.4% (253/307例) |
80.5% (252/313例) |
79.5% (244/307例) |
| ≧350 | 90.3% (130/144例) |
82.9% (136/164例) |
85.4% (123/144例) |
78.7% (129/164例) |
| ベースラインCD4陽性リンパ球数(cells/μL)別ウイルス学的失敗 | ||||
| <50 | 17.6% (6/34例) |
2.8% (1/36例) |
17.6% (6/34例) |
11.1% (4/36例) |
| ≧50、<200 | 13.9% (27/194例) |
8.0% (14/175例) |
19.1% (37/194例) |
8.0% (14/175例) |
| ≧200、<350 | 6.7% (21/313例) |
4.6% (14/307例) |
8.3% (26/313例) |
4.9% (15/307例) |
| ≧350 | 5.6% (8/144例) |
2.4% (4/164例) |
6.9% (10/144例) |
4.3% (7/164例) |
| BR別ウイルス学的効果 | ||||
| TDF/FTC | 83.5% (459/550例) |
82.4% (450/546例) |
76.9% (423/550例) |
77.3% (422/546例) |
| AZT/3TC | 87.1% (88/101例) |
80.6% (83/103例) |
81.2% (82/101例) |
76.7% (79/103例) |
| ABC/3TC | 88.6% (31/35例) |
84.8% (28/33例) |
77.1% (27/35例) |
84.8% (28/33例) |
注1)TLOVRアルゴリズム
注2)ウイルス学的再燃例(2回連続でHIV-1 RNA量<50copies/mLが認められ、その後48週又は96週までに2回連続でHIV-1 RNA量≧50copies/mLが認められた患者)又はウイルス学的非抑制例(48週又は96週までに2回連続したHIV-1 RNA量<50copies/mLが認められなかった患者)を含む。
48週時のCD4陽性リンパ球数のベースラインからの増加量の平均値はRPV+BR群で192cells/μL、EFV+BR群で176cells/μLであった[推定された投与群間差は17.9(95%信頼区間2.1~33.6)]。また、96週時のCD4陽性リンパ球数のベースラインからの増加量の平均値はRPV+BR群で228cells/μL、EFV+BR群で219cells/μLであった[推定された投与群間差は11.3(95%信頼区間-6.8~29.4)]。
17.1.2 抗HIV薬の使用経験のないHIV-1感染患者を対象とした海外臨床試験(第IIb相試験):C204試験
抗HIV薬の使用経験のないHIV-1感染患者368例を対象とし、3用量のRPV(25mg、75mg、150mg)及びBRの1日1回投与とEFV 600mg及びBRの1日1回投与の無作為割付け、実薬対照による臨床第IIb相比較試験を実施した。96週時までを用量設定相(RPV投与群のみ盲検化)、96週以降を長期投与相(非盲検)とした。BRはAZT/3TC、TDF/FTCから医師がBRを選択した。96週までのウイルス学的効果(HIV RNA量<50copies/mL)を表3に、96週以降240週までのウイルス学的効果を表4に示す。96週時におけるウイルス学的効果が認められた被験者の割合はRPV 25mg+BR群(N=93)で76.3%、EFV+BR群(N=89)で70.8%であった。CD4陽性リンパ球数のベースラインからの増加量の平均値はRPV 25mg+BR群で146cells/μL、EFV+BR群で160cells/μLであった。96週時においてウイルス学的効果が認められた被験者のうち、RPV+BR群では74%、EFV+BR群では81%が、240週時もウイルス学的効果を維持していた。34)[5.2 参照],[5.3 参照]
| RPV 25mg+BR群 N=93 |
EFV+BR群 N=89 |
|
|---|---|---|
| 16週 | 64例(68.8%) | 70例(78.7%) |
| 32週 | 73例(78.5%) | 76例(85.4%) |
| 48週 | 74例(79.6%) | 72例(80.9%) |
| 64週 | 72例(77.4%) | 69例(77.5%) |
| 80週 | 71例(76.3%) | 64例(71.9%) |
| 96週 | 71例(76.3%) | 63例(70.8%) |
注1)TLOVRアルゴリズム
| RPV+BR群注2) N=279 |
EFV+BR群 N=89 |
|
|---|---|---|
| 96週 | 204例(73.1%) | 63例(70.8%) |
| 120週 | 187例(67.0%) | 59例(66.3%) |
| 144週 | 180例(64.5%) | 55例(61.8%) |
| 168週 | 173例(62.0%) | 54例(60.7%) |
| 192週 | 163例(58.4%) | 54例(60.7%) |
| 216週 | 156例(55.9%) | 53例(59.6%) |
| 240週 | 152例(54.5%) | 51例(57.3%) |
注1)TLOVRアルゴリズム
注2)RPV+BRを投与している被験者は96週からRPVの用量を75mgとした。更に144週前後の時点からRPVの用量を25mgに変更した。
本剤25mgが投与された93例の安全性評価を行った。本剤の有害事象は90.3%(84/93例)に認められた。主な有害事象は、悪心31例(33.3%)、上気道感染17例(18.3%)、頭痛16例(17.2%)、浮動性めまい12例(12.9%)、単純ヘルペス10例(10.8%)等であった。
〈カボテグラビル経口剤併用時〉
17.1.3 国際共同第III相試験(FLAIR:201584試験)
抗レトロウイルス療法による治療経験のない成人HIV-1感染症患者を対象にインテグラーゼ阻害剤(INSTI)を含む1日1回1錠のレジメンからリルピビリンとカボテグラビルの併用療法に切り替えた後のウイルス学的抑制の維持の評価を目的としたランダム化非盲検比較試験に629例が組み入れられた。組み入れられた被験者にドルテグラビル・アバカビル・ラミブジン配合錠[HLA-B*5701陽性被験者では、ドルテグラビルと核酸系逆転写酵素阻害剤(NRTI)2剤]を1日1回、20週間経口投与した。HIV-1 RNA量が50copies/mL未満であった被験者566例(日本人患者20例を含む)のうち、リルピビリンとカボテグラビルの併用投与群(RPV+CAB群)に283例、ドルテグラビル・アバカビル・ラミブジン配合錠[HLA-B*5701陽性被験者では、ドルテグラビルと核酸系逆転写酵素阻害剤(NRTI)2剤]を継続する群(継続投与群)に283例が割り付けられた。RPV+CAB群に割り付けられた被験者には、リルピビリン経口剤25mgとカボテグラビル経口剤30mgを1日1回、少なくとも4週間併用経口投与した後、リルピビリン注射剤(1ヵ月目900mg、2ヵ月目以降600mg)とカボテグラビル注射剤(1ヵ月目600mg、2ヵ月目以降400mg)を1ヵ月間隔で44週間臀部筋肉内に併用投与した28)。両群の患者背景及び疾患特性に偏りはみられずRPV+CAB群の年齢中央値は34歳(範囲19-68歳)、女性22%、人種は白人76%、黒人又はアフリカ系アメリカ人17%、アジア人4%、その他が3%であった。ベースラインのCD4陽性リンパ球数350cells/mm3未満は7%であった。
主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、継続投与群の2.5%に対して、RPV+CAB群で2.1%であり、調整した群間差の95%信頼区間の上限値(2.1%)は、非劣性マージン(6%)より小さく、継続投与群に対するRPV+CAB群の非劣性が示された。48週時までにウイルス学的失敗の基準(HIV-1 RNA量が200copies/mL未満に抑制された後、2回の連続するHIV-1 RNA量の測定結果が200copies/mL以上)を満たした被験者はRPV+CAB群で1.4%(4/283例)、継続投与群で1.1%(3/283例)であった。48週時のベースライン特性別のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、RPV+CAB群及び継続投与群で同程度であった。日本人集団における主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者は、RPV+CAB群(8例)及び継続投与群(12例)両群ともに0例であった。[5.4 参照]
副作用発現頻度は、RPV+CAB群で83%(236/283例)であった。主な副作用は、注射部位疼痛78%(221/283例)、注射部位結節15%(43/283例)、注射部位硬結13%(37/283例)、注射部位腫脹8%(22/283例)、注射部位そう痒感6%(16/283例)、頭痛5%(14/283例)、発熱5%(13/283例)、注射部位紅斑4%(12/283例)、注射部位熱感3%(8/283例)及び体温上昇3%(8/283例)であった。日本人集団において2例以上にみられた副作用は、注射部位疼痛88%(7/8例)、倦怠感38%(3/8例)であった。
なお、本試験における試験成績の要約を表5に、ベースラインの特性別の48週時のHIV-1 RNA量が50copies/mL以上の被験者の割合を表6に示した。
| RPV+CAB群 283例 |
継続投与群 283例 |
|
|---|---|---|
| HIV-1 RNA量が50copies/mL以上注1) | 6例(2.1%) | 7例(2.5%) |
| 両群間の差(95%信頼区間)注2) | -0.4%(-2.8%, 2.1%) | |
| ウイルス学的失敗注3) | 4例(1.4%)注4) | 3例(1.1%) |
| RPV+CAB群 283例 |
継続投与群 283例 |
|
|---|---|---|
| ベースラインCD4陽性リンパ球数(cells/mm3) | ||
| <350 | 0/19 | 1/27(3.7%) |
| ≥350 to <500 | 3/64(4.7%) | 0/60 |
| ≥500 | 3/200(1.5%) | 6/196(3.1%) |
| 性別 | ||
| 男性 | 3/220(1.4%) | 6/219(2.7%) |
| 女性 | 3/63(4.8%) | 1/64(1.6%) |
| 人種 | ||
| 白人 | 6/216(2.8%) | 5/201(2.5%) |
| 黒人/アフリカ系米国人 | 0/47 | 2/56(3.6%) |
| アジア人 | 0/12 | 0/15 |
| その他 | 0/8 | 0/9 |
| BMI(kg/m2) | ||
| <30 | 3/243(1.2%) | 7/246(2.8%) |
| ≥30 | 3/40(7.5%) | 0/37 |
| 年齢(歳) | ||
| <50 | 5/250(2.0%) | 6/254(2.4%) |
| ≥50 | 1/33(3.0%) | 1/29(3.4%) |
17.1.4 海外第III相試験(ATLAS:201585試験)
抗レトロウイルス療法により、少なくとも6ヵ月間ウイルス学的に抑制されている成人HIV-1感染症患者616例を対象としたランダム化非盲検試験において、リルピビリンとカボテグラビルの併用投与群(RPV+CAB群)に308例、現行のレジメンを継続する群(継続投与群)に308例が割り付けられた。RPV+CAB群に割り付けられた被験者には、リルピビリン経口剤25mgとカボテグラビル経口剤30mgを1日1回、少なくとも4週間併用経口投与した後、リルピビリン注射剤(1ヵ月目900mg、2ヵ月目以降600mg)とカボテグラビル注射剤(1ヵ月目600mg、2ヵ月目以降400mg)を1ヵ月間隔で44週間臀部筋肉内に併用投与した29)。両群の患者背景及び疾患特性に偏りはみられずRPV+CAB群の年齢中央値は40歳(範囲21-74歳)、女性32%、人種は白人69%、黒人又はアフリカ系アメリカ人20%、アジア人7%、その他が3%であった。ベースラインのCD4陽性リンパ球数350cells/mm3未満は7%であった。
主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、継続投与群の1.0%に対して、RPV+CAB群で1.6%であり、調整した群間差の95%信頼区間の上限値(2.5%)は、非劣性マージン(6%)より小さく、継続投与群に対するRPV+CAB群の非劣性が示された。48週時までにウイルス学的失敗の基準(HIV-1 RNA量が200copies/mL未満に抑制された後、2回の連続するHIV-1 RNA量の測定結果が200copies/mL以上)を満たした被験者はRPV+CAB群で1.0%(3/308例)、継続投与群で1.3%(4/308例)であった。48週時のベースライン特性別のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、RPV+CAB群及び継続投与群で同程度であった。[5.4 参照]
副作用発現頻度は、RPV+CAB群で83%(255/308例)であった。主な副作用は、注射部位疼痛74%(227/308例)、注射部位結節12%(36/308例)、注射部位硬結9%(29/308例)、注射部位腫脹7%(22/308例)、注射部位紅斑4%(12/308例)、疲労4%(11/308例)、発熱4%(11/308例)、注射部位内出血3%(10/308例)、悪心4%(11/308例)、頭痛4%(11/308例)及び不眠症3%(8/308例)であった。
なお、本試験における試験成績の要約を表7に、ベースラインの特性別の48週時のHIV-1 RNA量が50copies/mL以上の被験者の割合を表8に示した。
| RPV+CAB群 308例 |
継続投与群 308例 |
|
|---|---|---|
| HIV-1 RNA量が50copies/mL以上注1) | 5例(1.6%) | 3例(1.0%) |
| 両群間の差(95%信頼区間)注2) | 0.6%(-1.2%, 2.5%) | |
| ウイルス学的失敗注3) | 3例(1.0%)注4) | 4例(1.3%) |
| RPV+CAB群 308例 |
継続投与群 308例 |
|
|---|---|---|
| ベースラインCD4陽性リンパ球数(cells/mm3) | ||
| <350 | 0/23 | 1/27(3.7%) |
| ≥350 to <500 | 2/56(3.6%) | 0/57 |
| ≥500 | 3/229(1.3%) | 2/224(0.9%) |
| 性別 | ||
| 男性 | 3/209(1.4%) | 3/204(1.5%) |
| 女性 | 2/99(2.0%) | 0/104 |
| 人種 | ||
| 白人 | 3/214(1.4%) | 2/207(1.0%) |
| 黒人/アフリカ系米国人 | 2/62(3.2%) | 1/77(1.3%) |
| アジア人 | 0/22 | 0/13 |
| その他 | 0/10 | 0/11 |
| BMI(kg/m2) | ||
| <30 | 3/248(1.2%) | 1/242(0.4%) |
| ≥30 | 2/60(3.3%) | 2/66(3.0%) |
| 年齢(歳) | ||
| <50 | 4/242(1.7%) | 2/212(0.9%) |
| ≥50 | 1/66(1.5%) | 1/96(1.0%) |
| ランダム化時の継続投与 | ||
| PI | 1/51(2.0%) | 0/54 |
| INSTI | 0/102 | 2/99(2.0%) |
| NNRTI | 4/155(2.6%) | 1/155(0.6%) |
17.1.5 海外第III相試験(ATLAS-2M:207966試験)
抗レトロウイルス療法により、ウイルス学的に抑制されている成人HIV-1感染症患者1045例を対象としたランダム化非盲検試験において、リルピビリンとカボテグラビルを1ヵ月間隔で併用投与する群(1ヵ月間隔投与群)に523例、2ヵ月間隔で併用投与する群(2ヵ月間隔投与群)に522例が割り付けられた。
割付け前にリルピビリンとカボテグラビルの併用療法以外の治療を受けていた被験者には、リルピビリン経口剤25mgとカボテグラビル経口剤30mgを1日1回、少なくとも4週間併用経口投与した。1ヵ月間隔投与群では、リルピビリン注射剤(1ヵ月目900mg、2ヵ月目以降1ヵ月間隔で600mg)とカボテグラビル注射剤(1ヵ月目600mg、2ヵ月目以降1ヵ月間隔で400mg)を44週間臀部筋肉内に併用投与した。2ヵ月間隔投与群では、リルピビリン注射剤(1、2ヵ月目及び以降2ヵ月間隔で900mg)とカボテグラビル注射剤(1、2ヵ月目及び以降2ヵ月間隔で600mg)を44週間臀部筋肉内に併用投与した30)。1ヵ月間隔投与群及び2ヵ月間隔投与群の患者背景及び疾患特性に偏りはみられず、年齢の中央値はいずれも42.0歳、性別は両群ともに男性が70%以上で、人種も70%以上が白人であり、CD4陽性リンパ球数350cells/mm3未満は、それぞれ5%及び7%であった。
主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、1ヵ月間隔投与群の1.0%に対して、2ヵ月間隔投与群で1.7%であり、調整した群間差の95%信頼区間の上限値(2.2%)は、非劣性マージン(4%)より小さく、1ヵ月間隔投与群に対する2ヵ月間隔投与群の非劣性が示された。48週時までにウイルス学的失敗の基準(HIV-1 RNA量が200copies/mL未満に抑制された後、2回の連続するHIV-1 RNA量の測定結果が200copies/mL以上)を満たした被験者は1ヵ月間隔投与群で0.4%(2/523例)、2ヵ月間隔投与群で1.5%(8/522例)であった。48週時のベースライン特性別のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、両群で同程度であった。[5.4 参照]
副作用発現頻度は、1ヵ月間隔投与群で76%(399/523例)、2ヵ月間隔投与群で77%(400/522例)であった。1ヵ月間隔投与群の主な副作用は、注射部位疼痛68%(358/523例)、注射部位結節17%(87/523例)、注射部位硬結7%(37/523例)、注射部位不快感8%(40/523例)、注射部位腫脹5%(26/523例)、発熱5%(25/523例)、注射部位そう痒感5%(24/523例)、疲労4%(19/523例)、注射部位紅斑3%(15/523例)及び注射部位血腫3%(14/523例)であり、2ヵ月間隔投与群の主な副作用は、注射部位疼痛70%(364/522例)、注射部位結節10%(54/522例)、注射部位硬結8%(40/522例)、注射部位不快感7%(34/522例)、注射部位腫脹6%(32/522例)、注射部位そう痒感5%(26/522例)及び発熱4%(19/522例)であった。
なお、本試験における試験成績の要約を表9に、ベースラインの特性別の48週時のHIV-1 RNA量が50copies/mL以上の被験者の割合を表10に示した。
| 1ヵ月間隔投与群 523例 |
2ヵ月間隔投与群 522例 |
|
|---|---|---|
| HIV-1 RNA量が50copies/mL以上注1) | 5例(1.0%) | 9例(1.7%) |
| 両群間の差(95%信頼区間)注2) | 0.8%(-0.6%, 2.2%) | |
| ウイルス学的失敗注3) | 2例(0.4%)注4) | 8例(1.5%)注4) |
| 1ヵ月間隔投与群 523例 |
2ヵ月間隔投与群 522例 |
|
|---|---|---|
| ベースラインCD4陽性リンパ球数(cells/mm3) | ||
| <350 | 1/27(3.7%) | 1/35(2.9%) |
| ≥350 to <500 | 0/89 | 1/96(1.0%) |
| ≥500 | 4/407(1.0%) | 7/391(1.8%) |
| 性別 | ||
| 男性 | 5/380(1.3%) | 4/385(1.0%) |
| 女性 | 0/143 | 5/137(3.6%) |
| 人種 | ||
| 白人 | 5/393(1.3%) | 5/370(1.4%) |
| 非白人 | 0/130 | 4/152(2.6%) |
| 黒人/アフリカ系米国人 | 0/90 | 4/101(4.0%) |
| 非黒人/アフリカ系米国人 | 5/433(1.2%) | 5/421(1.2%) |
| BMI(kg/m2) | ||
| <30 | 3/425(0.7%) | 3/409(0.7%) |
| ≥30 | 2/98(2.0%) | 6/113(5.3%) |
| 年齢(歳) | ||
| <35 | 1/145(0.7%) | 4/137(2.9%) |
| 35 to <50 | 2/239(0.8%) | 3/242(1.2%) |
| ≥50 | 2/139(1.4%) | 2/143(1.4%) |
| RPV+CAB投与歴(週) | ||
| None | 5/327(1.5%) | 5/327(1.5%) |
| 1-24 | 0/68 | 3/69(4.3%) |
| >24 | 0/128 | 1/126(0.8%) |
17.3 その他
17.3.1 QT間隔に対する影響
健康成人60例を対象に本剤25mg(臨床用量)を1日1回反復経口投与し、本剤の定常状態時のQTcF間隔に及ぼす影響を検討した結果、QTcF間隔に対し臨床的に有意な影響は認められなかった(プラセボとの差の最大値:2.2ms)[プラセボ及び陽性対照(moxifloxacin 400mg 1日1回)を用いた無作為割付クロスオーバー試験]。
なお、健康成人におけるQT/QTc評価試験において、高用量のリルピビリン(75mg及び300mg)注)を1日1回反復経口投与したとき、QTcF間隔のベースラインからの変化量のプラセボとの差の平均値(95%信頼区間の上限)はそれぞれ10.7(15.3)ms及び23.3(28.4)msであった。31),32) (外国人データ)[9.1.1 参照],[10.2 参照]
18. 薬効薬理
18.1 作用機序
リルピビリンはジアリルピリミジン骨格を有し、HIV-1に作用するNNRTIである。リルピビリンは、HIV-1逆転写酵素(RT)を非競合的に阻害し、ヒトDNAポリメラーゼα、β及びγを阻害しない。35),36)
18.2 抗ウイルス作用
T細胞株に急性感染させた野生型(WT)HIV-1実験室株のIIIBに対するリルピビリンの50%有効濃度(EC50)の中央値は、0.73nmol/L(0.27ng/mL)であった。
リルピビリンはHIV-1臨床分離株のgroup Mに対して0.07~1.01nmol/L(0.03~0.37ng/mL)、group Oに対して2.88~8.45nmol/L(1.06~3.10ng/mL)のEC50値を示した。
リルピビリンは、NRTI/NtRTI(アバカビル、ジダノシン、エムトリシタビン、サニルブジン及びテノホビル)、プロテアーゼ阻害剤(アンプレナビル、アタザナビル、ダルナビル、インジナビル、ロピナビル、ネルフィナビル、リトナビル、サキナビル及びtipranavir)、NNRTI(エファビレンツ、エトラビリン及びネビラピン)、融合阻害剤(enfuvirtide)及びCCR5阻害剤(マラビロク)との併用により相加作用を示した。NRTIであるラミブジン及びジドブジン、インテグラーゼ阻害剤であるラルテグラビルとは相加又は相乗作用を示した。35)
18.3 薬剤耐性
異なる由来及びサブタイプのWT又はNNRTI耐性HIV-1株を用いたin vitro耐性獲得試験において、リルピビリン耐性株が出現した。この耐性株で最も高頻度で出現したアミノ酸変異はL100I、K101E、V108I、E138K、V179F、Y181C、H221Y、F227C及びM230Iであった。
生物学的カットオフ値(BCO)を超えるFC値[表現型耐性の指標であるEC50値の比(各種分離株に対するEC50値/WT HIV-1株に対するEC50値)]を示した株を、リルピビリン耐性とした。
第III相試験の48週時併合解析において、本剤投与群のウイルス学的失敗例72例のうち62例にベースライン時からの耐性変異が認められた。NNRTI耐性を示すアミノ酸変異は主に、V90I、L100I、K101E、E138K、E138Q、V179I、Y181C、V189I、H221Y及びF227Cが認められた。48週時に認められた変異は96週時にも認められた。ベースライン時にみられたV90I及びV189Iは本試験で効果に影響を及ぼさなかった。リルピビリン投与期間にE138Kのアミノ酸変異が最も高い頻度で発現し、多くがM184Iのアミノ酸変異を伴っていた。
In vitro及びin vivoでの検討結果から、ベースライン時にK101E、K101P、E138A、E138G、E138K、E138R、E138Q、V179L、Y181C、Y181I、Y181V、Y188L、H221Y、F227C、M230I及びM230Lのアミノ酸変異を有する株は、リルピビリンの抗ウイルス効果に影響を及ぼす可能性があると考えられた。33),34),35),37)
18.4 交差耐性
リルピビリンは、RTにK103N及びY181C等のNNRTI耐性関連アミノ酸変異を1個導入した67株のうち64株(96%)に抗ウイルス作用を示した。リルピビリンへの感受性の低下をもたらした単一のアミノ酸変異はK101P、Y181I及びY181Vであった。K103Nのアミノ酸変異は、単一でリルピビリンに対する感受性が低下しなかったが、K103N及びL100Iの二重変異では、リルピビリンに対する感受性が7倍低下した。
エファビレンツ及びネビラピンのどちらか一方若しくは両方に耐性を示す4786株のHIV-1組換え型臨床分離株のうち62%の株は、リルピビリンに対して感受性を維持(FC値≦BCO)していた。
第III相試験の48週時併合解析において、RPV+BR群のウイルス学的失敗62例中31例が表現型耐性検査にて本剤に対する感受性が低下していた。これらのうち28例はエトラビリン、27例はエファビレンツ、14例はネビラピンへの耐性を示した。48週時に認められた交差耐性は96週時にも認められた。
第III相試験の96週時併合解析において、本剤に耐性を示したRPV+BR群のウイルス学的失敗例の中では、ベースラインHIV RNA量が>100,000copies/mLの被験者よりもベースラインHIV RNA量が≦100,000copies/mLの被験者の方が、表現型交差耐性を示した被験者数は少なかった。本剤に耐性を示すウイルス学的失敗例において、ベースラインのHIV RNA量が≦100,000copies/mLの被験者5例のうち、3例はエファビレンツ、4例はエトラビリン、1例はネビラピンへの耐性を示した。ベースラインのHIV RNA量が>100,000copies/mLの被験者30例のうち、27例はエファビレンツ、28例はエトラビリン、15例はネビラピンへの耐性を示した。33),35),38)
19. 有効成分に関する理化学的知見
| 一般的名称 | リルピビリン塩酸塩(Rilpivirine Hydrochloride) |
|---|---|
| 化学名 | 4-{[4-({4-[(1E)-2-Cyanoethenyl]-2,6-dimethylphenyl}amino)pyrimidin-2-yl]amino}benzonitrile monohydrochloride |
| 分子式 | C22H18N6・HCl |
| 分子量 | 402.88 |
| 性状 | 白色の粉末 |
| 化学構造式 | 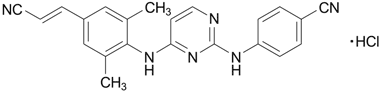 |
| 融点 | 約250℃(分解) |
| 分配係数 | log P=4.86(1-オクタノール/pH 7.0リン酸緩衝液) |
| 溶解性 | メタノール 5.8mg/mL エタノール 0.67mg/mL 水 0.01mg/mL |
20. 取扱い上の注意
20.1
本剤は遮光保存する必要があるため、服用直前にボトルから取り出すよう指導すること。
22. 包装
30錠[ボトル、バラ]
23. 主要文献
- 社内資料:リファンピシンとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.1)
- 社内資料:オメプラゾールとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.9)
- 社内資料:リファブチンとリルピビリンの相互作用[TMC278IFD1003]
- 社内資料:ファモチジンとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.10)
- 社内資料:メサドンとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.8)
- 社内資料:ジダノシンとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.1.2)
- 社内資料:テノホビルとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.1.1)
- 社内資料:ダルナビル/リトナビルとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.1.4)
- 社内資料:ロピナビル/リトナビルとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.1.3)
- 社内資料:ラルテグラビルとリルピビリンの相互作用[TMC278-C153]
- 社内資料:リルピビリンの薬物動態の検討[TMC278-IFD4005]
- 社内資料:リルピビリンの薬物動態の検討(2012年5月18日承認、CTD2.7.1-2.2.1)
- 社内資料:リルピビリンの薬物動態に及ぼす食事の影響の検討(2012年5月18日承認、CTD2.7.1-2.3.1)
- 社内資料:リルピビリンの蛋白結合に関する検討(2012年5月18日承認、CTD2.7.2-2.1.2)
- 社内資料:リルピビリンの代謝に関する検討(2012年5月18日承認、CTD2.7.2-2.1.3.3)
- 社内資料:リルピビリンの薬物動態の検討(2012年5月18日承認、CTD2.7.2-2.2)
- 社内資料:リルピビリンの薬物動態の検討(2012年5月18日承認、CTD2.7.2-2.7)
- 社内資料:リファブチンとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.2)
- 社内資料:ケトコナゾールとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.3)
- 社内資料:アセトアミノフェンとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.11)
- 社内資料:アトルバスタチンとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.5)
- 社内資料:クロルゾキサゾンとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.12)
- 社内資料:シルデナフィルとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.4)
- 社内資料:カボテグラビルとリルピビリンの相互作用[LAI116181]
- 社内資料:エチニルエストラジオール/ノルエチステロンとリルピビリンの相互作用(2012年5月18日承認、CTD2.7.2-2.8.3.7)
- 社内資料:メトホルミンとリルピビリンの相互作用[TMC278IFD1004]
- 社内資料:ジゴキシンとリルピビリンの相互作用[TMC278IFD1001]
- Orkin C, et al.:N Engl J Med. 2020; 382 (12):1124-1135
- Swindells S, et al.:N Engl J Med. 2020; 382 (12):1112-1123
- Overton ET, et al.:Lancet. 2020; 396:1994-2005
- 社内資料:リルピビリンのQT間隔に対する作用(2012年5月18日承認、CTD2.7.2-2.9.3)
- 社内資料:リルピビリンのQT間隔に対する作用(2012年5月18日承認、CTD2.7.2-2.9.1)
- 社内資料:リルピビリンとエファビレンツの初回治療HIV-1感染患者に対する臨床成績[TMC278-C904](2012年5月18日承認、CTD2.7.3-3.1)
- 社内資料:リルピビリンの初回治療HIV-1感染患者に対する臨床成績(2012年5月18日承認、CTD2.7.3-2.3)
- Azijn H, et al.:Antimicrob Agents Chemother. 2010; 54: 718-727
- 社内資料:リルピビリンの作用機序(2012年5月18日承認、CTD2.6.2-3.2)
- Mojgan H, et al.:19th Conference on Retroviruses and Opportunistic Infection. 2012; March 5-8
- 社内資料:リルピビリンの抗ウイルス作用(2012年5月18日承認、CTD2.7.2-3.2.3)
24. 文献請求先及び問い合わせ先
ヤンセンファーマ株式会社 メディカルインフォメーションセンター
〒101-0065 東京都千代田区西神田3-5-2
フリーダイヤル 0120-183-275
https://www.janssenpro.jp
26. 製造販売業者等
26.1 製造販売元
ヤンセンファーマ株式会社
〒101-0065 東京都千代田区西神田3-5-2