アイセントレス錠400mgの添付文書
- 商品名:
- アイセントレス錠400mg
- 一般名:
- ラルテグラビル
- 略称 :
- RAL

添付文書の読み方
ここで提供している添付文書情報は、2023年11月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
HIVインテグラーゼ阻害剤
ラルテグラビルカリウム錠
- **2023年8月改訂(第3版)
- *2021年5月改訂(第2版)
劇薬
処方箋医薬品:注意―医師等の処方箋により使用すること
| 日本標準商品分類番号 | 87625 |
|---|
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 3年 |
| 承認番号 | 22000AMX01647000 |
|---|---|
| 販売開始 | 2008年7月 |
2. 禁忌(次の患者には投与しないこと)
2.1
本剤の成分に対し過敏症の既往歴のある患者
3. 組成・性状
3.1 組成
| 有効成分 | 1錠中ラルテグラビルカリウム 434.4mg(ラルテグラビルとして 400mg) |
|---|---|
| 添加剤 | 結晶セルロース、乳糖水和物、無水リン酸水素カルシウム、ヒプロメロース、ポリオキシエチレン(196)ポリオキシプロピレン(67)グリコール、フマル酸ステアリルナトリウム、ステアリン酸マグネシウム、ポリビニルアルコール(部分けん化物)、マクロゴール4000、タルク、酸化チタン、三二酸化鉄、黒酸化鉄 |
3.2 製剤の性状
| 剤形 | 楕円形・フィルムコーティング錠 | |
|---|---|---|
| 色調 | うすい赤色 | |
| 外形 | 表面 |  |
| 裏面 |  |
|
| 側面 |  |
|
| 大きさ | 長径 | 15.9mm |
| 短径 | 8.8mm | |
| 厚さ | 7.0mm | |
| 識別コード |  227 227 |
|
4. 効能又は効果
HIV感染症
5. 効能又は効果に関連する注意
本剤による治療にあたっては、患者の治療歴及び薬剤耐性検査結果を参考にすること。
6. 用法及び用量
通常、成人にはラルテグラビルとして400mgを1日2回経口投与する。本剤は、食事の有無にかかわらず投与できる。なお、投与に際しては、必ず他の抗HIV薬と併用すること。
8. 重要な基本的注意
**,*8.1
本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又はそれに代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。
- 本剤はHIV感染症の根治療法薬ではないことから、日和見感染を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。
- 本剤の長期投与による影響については、現在のところ不明である。
- 本剤の抗ウイルス効果を最大にするために、担当医の指示なしに用量を変更したり、服用を中止したりしないこと。
8.2
本剤を含む抗HIV薬の多剤併用療法を行った患者で、免疫再構築症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
9. 特定の背景を有する患者に関する注意
9.5 妊婦
妊婦又は妊娠している可能性のある女性には治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
ラット及びウサギにおける高用量投与で、胎盤移行が認められている1)。また、ラットにおける高用量投与で、過剰肋骨が報告されている。
9.6 授乳婦
授乳を避けさせること。
動物実験(ラット)で乳汁中へ移行することが報告されている2)。ラルテグラビルがヒトの乳汁中に移行するか否かは不明である。乳汁を介してHIV母児感染の可能性がある。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
一般に、肝、腎又は心機能が低下し、合併症を有している又は他の薬剤を併用している場合が多い。
10. 相互作用
ラルテグラビルは、主にUDP-グルクロノシルトランスフェラーゼ(UGT)1A1によるグルクロン酸抱合によって代謝される。[16.4 参照],[16.7.3 参照]
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
|
本剤投与前後6時間以内に水酸化マグネシウム・水酸化アルミニウム含有制酸剤を併用投与した場合、本剤の血漿中濃度が低下する。 | これらの薬剤とのキレート形成による本剤の吸収抑制等がおこるおそれがある。 |
|
本剤投与前後6時間以内に水酸化マグネシウム・水酸化アルミニウム含有制酸剤を併用投与した場合、本剤の血漿中濃度が低下する。 | これらの薬剤とのキレート形成による本剤の吸収抑制等がおこるおそれがある。 |
11. 副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1 重大な副作用
11.1.1 皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)
11.1.2 薬剤性過敏症症候群(頻度不明)
初期症状として発疹、発熱がみられ、さらに肝機能障害、リンパ節腫脹、白血球増加、好酸球増多、異型リンパ球出現等を伴う遅発性の重篤な過敏症状があらわれることがある。なお、ヒトヘルペスウイルス6(HHV-6)等のウイルスの再活性化を伴うことが多く、投与中止後も発疹、発熱、肝機能障害等の症状が再燃あるいは遷延化することがあるので注意すること3)。
11.1.3 過敏症(頻度不明)
11.1.4 横紋筋融解症、ミオパチー(いずれも頻度不明)
筋肉痛、脱力感、CK上昇、血中及び尿中ミオグロビン上昇を特徴とする横紋筋融解症があらわれ、急性腎障害等の重篤な腎障害があらわれることがある。また、ミオパチーがあらわれることがあるので、筋力低下、筋痛や著明なCKの上昇があらわれた場合には投与を中止すること。
11.1.5 腎不全(0.1%)
11.1.6 肝炎(0.1%)
重篤な肝炎があらわれることがある。
11.1.7 胃炎(0.3%)
重篤な胃炎があらわれることがある。
11.1.8 陰部ヘルペス(0.1%)
重篤な陰部ヘルペスがあらわれることがある。
11.2 その他の副作用
| 2%以上 | 2%未満 | 頻度不明 | |
|---|---|---|---|
| 血液及びリンパ系障害 | 貧血、好中球減少症、リンパ節痛、リンパ節症 | 血小板減少症 | |
| 心臓障害 | 動悸、心室性期外収縮、洞性徐脈 | ||
| 耳及び迷路障害 | 回転性めまい、耳鳴 | ||
| 胃腸障害 | 下痢、悪心 | 腹痛、嘔吐、腹部膨満、便秘、腹部不快感、消化不良、鼓腸、舌炎、胃食道逆流性疾患、口内乾燥、おくび、びらん性十二指腸炎、腹部圧痛、唾液欠乏、歯肉炎 | |
| 肝胆道系障害 | 脂肪肝 | ||
| 全身障害及び投与局所様態 | 疲労 | 無力症、発熱、悪寒、熱感、顔面浮腫、末梢性浮腫、顎下腫瘤、疼痛 | |
| 感染症及び寄生虫症 | 単純ヘルペス、帯状疱疹、胃腸炎、毛包炎、リンパ節膿瘍、鼻咽頭炎、上気道感染 | ||
| 代謝及び栄養障害 | 体脂肪の再分布/蓄積(脂肪組織萎縮症、脂肪肥大症、顔のやせ、中心性肥満、異脂肪血症) | 糖尿病、食欲亢進、食欲減退、過食、多飲症 | |
| 筋骨格系及び結合組織障害 | 関節痛、筋痛、背部痛、筋骨格痛、筋萎縮症、骨粗鬆症、関節炎、頚部痛、多発性関節炎、側腹部痛、骨減少症、四肢痛 | ||
| 神経系障害 | 頭痛、浮動性めまい | ニューロパチー、錯感覚、傾眠、緊張性頭痛、振戦、記憶障害、認知障害、注意力障害、感覚鈍麻、睡眠の質低下、片頭痛 | 小脳性運動失調 |
| 精神障害 | 不眠症、異常な夢 | うつ病、不安、錯乱状態、気分変化、パニック発作、睡眠障害 | 自殺企図 |
| 腎及び尿路障害 | 腎炎、間質性腎炎、腎結石症、頻尿、腎嚢胞 | ||
| 生殖系及び乳房障害 | 勃起不全、女性化乳房 | ||
| 皮膚及び皮下組織障害 | 発疹、多汗症、紅斑、寝汗、乾皮症、痒疹、ざ瘡、脱毛症、そう痒症、じん麻疹 | ||
| 臨床検査 | AST上昇、ALT上昇、総ビリルビン上昇、CK上昇 | ||
| その他 | 視覚障害、鼻出血、体重減少、体重増加 |
15. その他の注意
15.2 非臨床試験に基づく情報
1群あたり雌雄各50匹のラットに、それぞれラルテグラビル50(雌雄)、150(雄)、300(雌雄)又は600(雌)mg/kg/日を投与した長期(2年間)がん原性試験を実施したところ、300及び600mg/kg/日投与群で鼻/鼻咽頭の腫瘍(扁平上皮癌)が認められたが、これらの腫瘍は種特異的であると考えられる。なお、マウスがん原性試験においては、ラルテグラビルの発がん性は認められなかった。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
(1)
健康成人男性に対してラルテグラビル400mgを空腹時単回経口投与したところ、ラルテグラビルは速やかに吸収され、その後、二相性で消失した。
| AUC0-∞ (μM・hr) |
Cmax (μM) |
C12hr (nM) |
Tmax (hr) |
t1/2α (hr) |
t1/2β (hr) |
|---|---|---|---|---|---|
| 11.7 | 3.1 | 137.5 | 3.0 | 1.0 | 7.3 |
(2)
健康成人にラルテグラビルを空腹時単回投与したところ、速やかに吸収され、Tmaxは投与後約3時間であった。ラルテグラビルのAUC及びCmaxは、100~1,600mgの範囲で用量に比例して増加した。また、ラルテグラビルのC12hrは、100~800mgの範囲で用量に比例して増加したが、100~1,600mgの範囲では用量比例性をやや下回った4)。 ラルテグラビルの見かけの消失半減期は終末相では約9時間、α相ではより短い約1時間である。また、AUCに対するこのα相での消失の寄与は大きかった(外国人データ)。
16.1.2 反復投与
健康成人にラルテグラビル100~800mgを1日2回10日間反復投与したところ、投与開始からほぼ2日以内に定常状態に到達した。AUC及びCmaxの値から判断すると蓄積は殆どみられず、C12hrからみた蓄積もわずかであった4)(外国人データ)。
HIV感染患者にラルテグラビル400mgを1日2回10日間反復投与したところ、投与10日目におけるラルテグラビルのAUC0-12hr及びC12hrの幾何平均値はそれぞれ14.3μM・hr及び142nMであった5)(外国人データ)。
16.2 吸収
16.2.1 食事の影響
本剤は食事の有無にかかわらず投与できる。HIV感染患者を対象とした主要な安全性及び有効性試験では、ラルテグラビルを食事と関係なく投与した。定常状態におけるラルテグラビルの薬物動態に及ぼす低、中及び高脂肪食の摂取の影響について、健康被験者を対象に評価した。中脂肪食摂取後のラルテグラビル反復投与では、空腹時に比べてラルテグラビルのAUCが13%増加したが、臨床的には意味のない程度の変化であった。また、空腹時に比べて、ラルテグラビルのC12hrは66%高く、Cmaxは5%高かった。高脂肪食摂取後のラルテグラビル投与では、AUC及びCmaxは約2倍、C12hrは4.1倍増加した。低脂肪食摂取後のラルテグラビル投与では、AUC及びCmaxはそれぞれ46%及び52%減少したが、C12hrについては本質的な変化はみられなかった。空腹時に比べ、食事摂取によって本剤の薬物動態のばらつきが増大すると考えられる6)(外国人データ)。
16.2.2 生物学的利用率
ラルテグラビルの絶対生物学的利用率を求める試験は実施していない。
16.3 分布
ラルテグラビルのヒト血漿蛋白との結合率は、2~10μMの濃度範囲で約83%であった。
ラルテグラビルは、ラットにおいて容易に胎盤を通過したが、脳内移行性は低かった。
HIV-1感染患者にラルテグラビル400mgを1日2回投与した2つの試験で、ラルテグラビルは脳脊髄液中に検出された。各試験でのラルテグラビルの脳脊髄液中濃度(中央値)はそれぞれ血漿中濃度の5.8%(範囲:1%~53.5%)(n=18)及び3%(範囲:1%~61%)(n=16)に相当した7),8)。これらは血漿中遊離体濃度の約1/3~1/6倍の濃度であった(外国人データ)。
16.4 代謝
健康成人に放射能標識したラルテグラビルを経口投与したところ、尿中にはラルテグラビル及びそのグルクロン酸抱合体が検出され、それぞれ投与量の約9%及び23%に相当した。糞中にはラルテグラビルのみが存在し、その大部分は非臨床動物試験で認められたように胆汁中に排泄されたラルテグラビルのグルクロン酸抱合体が加水分解されて生成すると考えられる(外国人データ)。
血漿中の主要な成分はラルテグラビルであり、総放射能の約70%を占め、残りの放射能はラルテグラビルのグルクロン酸抱合体であった(外国人データ)。
酵素分子種に選択的な化学的阻害剤及びcDNA発現系UGTを用いた試験で、UGT1A1が、ラルテグラビルのグルクロン酸抱合体形成に関与する主要な酵素であることが示された。ヒトにおけるラルテグラビルの主要な消失機序は、UGT1A1を介するグルクロン酸抱合である9)(外国人データ)。[10. 参照]
16.5 排泄
健康成人に放射能標識したラルテグラビルを経口投与したところ、投与量の約32%及び51%がそれぞれ尿中及び糞中に排泄された9)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
未変化体の腎を介した排泄は、主要な消失経路ではない。重度腎機能障害患者においてラルテグラビルの薬物動態試験を実施した。また、統合薬物動態解析においても、腎機能障害の影響を評価した。重度腎機能障害患者と健康被験者間では、臨床的に重要な薬物動態の差はみられず(外国人データ)、用量調節の必要はない。血液透析によるラルテグラビル除去の程度は不明のため、透析実施前には本剤の投与を避けること10)。
16.6.2 肝機能障害患者
ラルテグラビルは、主として肝臓でのグルクロン酸抱合により消失する。中等度肝機能障害患者においてラルテグラビルの薬物動態試験を実施した。また、統合薬物動態解析においても、肝機能障害の影響を評価した。中等度肝機能障害患者と健康被験者間では、臨床的に重要な薬物動態の差はみられなかった(外国人データ)。軽度から中等度の肝機能障害患者では、用量調節の必要はない。ラルテグラビルの薬物動態に及ぼす重度肝機能障害の影響は検討されていない11)。
16.6.3 小児等
16歳未満の小児患者におけるラルテグラビルの薬物動態は確立していない。
16.6.4 その他の要因
(1) 性別
性別の影響について、空腹時投与におけるラルテグラビル単独療法を受けた健康被験者103例及びHIV感染患者28例の薬物動態データを用いた統合解析により評価した。また、性別の影響を、空腹時及び摂食後にラルテグラビル単独投与又は他剤との併用投与を受けた健康被験者及びHIV感染患者80例の濃度データに関する母集団薬物動態解析においても評価した。これらの解析において、性別に起因する臨床的に重要な薬物動態の差は認められなかった(外国人データ)。用量調節の必要はない。
(2) 年齢
統合解析及び母集団薬物動態解析において、年齢はラルテグラビルの薬物動態に臨床的に意味のある影響を及ぼさなかった(外国人データ)。用量調節の必要はない。
(3) 人種
統合解析において、人種はラルテグラビルの薬物動態に臨床的に意味のある影響を及ぼさなかった(外国人データ)。用量調節の必要はない。
(4) Body Mass Index(BMI)
統合解析において、BMIはラルテグラビルの薬物動態に臨床的に意味のある影響を及ぼさなかった。また、母集団薬物動態解析において、体重もラルテグラビルの薬物動態に臨床的に意味のある影響を及ぼさなかった(外国人データ)。用量調節の必要はない。
(5) UGT1A1遺伝多型
UGT1A1の遺伝多型によってラルテグラビルの薬物動態が臨床的に意味のある影響を受けるという証拠はない。*28/*28遺伝子型を持つ被験者30例と野生型の遺伝子型を持つ被験者27例との比較において、AUCの幾何平均比(90%信頼区間)は1.41(0.96, 2.09)であった12)(外国人データ)。
16.7 薬物相互作用
16.7.1
ラルテグラビルは、チトクロームP450(CYP)の基質ではなく、in vitroでCYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6又はCYP3Aを阻害しなかった(IC50>100μM)。さらに、in vitroでラルテグラビルはCYP3A4を誘導しなかった13)。CYP3A4の鋭敏な基質であるミダゾラムとの薬物相互作用試験において、ラルテグラビルはミダゾラムの薬物動態に有意な影響を及ぼさなかった14)(外国人データ)ことから、ラルテグラビルが、in vivoでCYP3A4によって代謝される薬剤の薬物動態を変化させる可能性は低い。
16.7.2
ラルテグラビルは、UGT1A1、UGT2B7を阻害することはなく(IC50>50μM)、またP-糖蛋白による輸送も阻害しないこと13)から、ラルテグラビルはこれらの酵素又はP-糖蛋白の基質である薬剤(例えば、プロテアーゼ阻害剤、非核酸系逆転写酵素阻害剤、メサドン、オピオイド系鎮痛薬、スタチン類、アゾール系抗真菌薬、プロトンポンプ阻害剤及び勃起不全治療薬)の薬物動態に影響を及ぼさないと予想される。
16.7.3
In vivo(外国人データ)及びin vitro試験によれば、ラルテグラビルは、主にUGT1A1によるグルクロン酸抱合を介する代謝によって消失する9)。フェニトイン又はフェノバルビタールのようなリファンピシン以外の強力な薬物代謝酵素誘導剤のUGT1A1に対する影響は不明である。他の作用の弱い誘導剤[例えば、エファビレンツ(EFV)、ネビラピン、リファブチン、グルココルチコイド、セント・ジョーンズ・ワート及びピオグリタゾン]は、本剤の推奨用量と併用することができる。[10. 参照],[10.2 参照]
16.7.4
ラルテグラビルと強力なUGT1A1阻害剤であることが知られている薬剤(例えば、アタザナビル15))との併用は、ラルテグラビルの血漿中濃度を増加させる可能性がある。しかしながら、増加の程度は大きくなく、また、これら阻害剤との併用療法は、臨床試験で良好な忍容性を示した(外国人データ)ことから、ラルテグラビルの用量調節の必要はない(表2)。
16.7.5
2価金属イオンを含む制酸剤と併用した場合、キレート形成による本剤の吸収抑制等がおこる可能性がある。本剤服用前後6時間以内の水酸化マグネシウム・水酸化アルミニウム含有制酸剤の服用により本剤のCminが著しく低下した(外国人データ)。[10.2 参照]
16.7.6
本剤の溶解度はより高いpHにおいて増大するため、胃内pHを上昇させることが知られている薬剤(例えば、オメプラゾール)と本剤との併用により、本剤の血漿中濃度が上昇する可能性がある。BENCHMRK 1及び2試験におけるプロトンポンプ阻害剤又はH2ブロッカーと本剤の併用投与例では、非併用例のサブグループと同様の安全性プロファイルが認められた(外国人データ)。これらのデータに基づき、プロトンポンプ阻害剤又はH2ブロッカーは、用量調整せず本剤と併用することができる16)。
16.7.7 薬物相互作用臨床試験
(1) 他剤の薬物動態に及ぼすラルテグラビルの影響
薬物相互作用試験において、ラルテグラビルは、ホルモン避妊薬17)、メサドン18)、TDF19)、ミダゾラム14)、ラミブジン20)及びエトラビリン21)の薬物動態に臨床的に意味のある影響を及ぼさなかった。反復投与薬物相互作用試験において、ラルテグラビルと併用した場合のエチニルエストラジオールとノルエルゲストロミンのAUCは、ラルテグラビル非併用時のそれぞれ98%及び114%であった17)。また、反復投与薬物相互作用試験において、ラルテグラビルと併用した場合のテノホビルのAUC及びトラフ濃度は、TDF単独投与時の90%及び87%であった19)。別の薬物相互作用試験において、ラルテグラビルと併用した場合のミダゾラムのAUCは、ミダゾラム単独投与時の92%であった14)。第II相試験において、ラミブジンの薬物動態は、ラルテグラビル併用群とEFV併用群で同様であった20)(外国人データ)。
(2) ラルテグラビルの薬物動態に及ぼす他剤の影響
薬物相互作用試験において、アタザナビル15)、EFV22)、リトナビル22)、TDF19)、tipranavir/リトナビル23)及び炭酸カルシウム含有制酸剤は、ラルテグラビルの薬物動態に臨床的に意味のある影響を及ぼさなかった。強力な薬物代謝酵素誘導剤であるリファンピシンは、ラルテグラビルのトラフ濃度の低下をもたらした。薬物相互作用の詳細を、表2に記載する(外国人データ)。[10.2 参照]
| 併用薬 | 併用薬 |
ラルテグラビル |
ラルテグラビルの薬物動態パラメータ比 併用時/非併用時 (90%信頼区間);影響なし=1.00 |
|||
|---|---|---|---|---|---|---|
| n | Cmax | AUC | Cmin | |||
| 水酸化アルミニウム・水酸化マグネシウム | 1600mg・1600mg 同時 単回投与 |
400mg 1日2回 |
25 | 0.56 (0.42, 0.73) |
0.51 (0.40, 0.65) |
0.37 (0.29, 0.48) |
| 1600mg・1600mg 2h前 単回投与 |
23 | 0.49 (0.33, 0.71) |
0.49 (0.35, 0.67) |
0.44 (0.34, 0.55) |
||
| 1600mg・1600mg 2h後 単回投与 |
23 | 0.78 (0.53, 1.13) |
0.70 (0.50, 0.96) |
0.43 (0.34, 0.55) |
||
| 1600mg・1600mg 6h前 単回投与 |
16 | 0.90 (0.58, 1.40) |
0.87 (0.64, 1.18) |
0.50 (0.39, 0.65) |
||
| 1600mg・1600mg 6h後 単回投与 |
16 | 0.90 (0.58, 1.41) |
0.89 (0.64, 1.22) |
0.51 (0.40, 0.64) |
||
| アタザナビル15) | 400mg 1日1回 |
100mg 単回投与 |
10 | 1.53 (1.11, 2.12) |
1.72 (1.47, 2.02) |
1.95 (1.30, 2.92) |
| アタザナビル リトナビル15) |
300mg 1日1回 100mg 1日1回 |
400mg 1日2回 |
10 | 1.24 (0.87, 1.77) |
1.41 (1.12, 1.78) |
1.77 (1.39, 2.25) |
| 炭酸カルシウム | 3000mg 単回投与 |
400mg 1日2回 |
24 | 0.48 (0.36, 0.63) |
0.45 (0.35, 0.57) |
0.68 (0.53, 0.87) |
| EFV22) | 600mg 1日1回 |
400mg 単回投与 |
9 | 0.64 (0.41, 0.98) |
0.64 (0.52, 0.80) |
0.79 (0.49, 1.28) |
| オメプラゾール16) | 20mg 1日1回 |
400mg 単回投与 |
14 | 4.15 (2.82, 6.10) |
3.12注1) (2.13, 4.56) |
1.46 (1.10, 1.93) |
| リファンピシン24) | 600mg 1日1回 |
400mg 単回投与 |
9 | 0.62 (0.37, 1.04) |
0.60 (0.39, 0.91) |
0.39 (0.30, 0.51) |
| リファンピシン24) | 600mg 1日1回 |
800mg 1日2回 |
14 | 1.62注3) (1.12, 2.33) |
1.27注3) (0.94, 1.71) |
0.47注3) (0.36, 0.61) |
| リトナビル22) | 100mg 1日2回 |
400mg 単回投与 |
10 | 0.76 (0.55, 1.04) |
0.84 (0.70, 1.01) |
0.99 (0.70, 1.40) |
| TDF19) | 300mg 1日1回 |
400mg 1日2回 |
9 | 1.64 (1.16, 2.32) |
1.49 (1.15, 1.94) |
1.03 (0.73, 1.45) |
| tipranavir リトナビル23) |
500mg 1日2回 200mg 1日2回 |
400mg 1日2回 |
15 | 0.82 (0.46, 1.46) |
0.76 (0.49, 1.19) |
0.45注2) (0.31, 0.66) |
|
||||||
注)本剤の承認された用法及び用量は400mgを1日2回である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 抗HIV治療経験患者を対象にした海外第III相臨床試験(BENCHMRK 1及び2)
BENCHMRK 1及び2(無作為化、二重盲検、プラセボ対照試験)は、3クラスの抗HIV薬[核酸系逆転写酵素阻害剤(NRTI)、非核酸系逆転写酵素阻害剤(NNRTI)、プロテアーゼ阻害剤(PI)]の各々で1剤以上に耐性が証明された、16歳以上のHIV感染患者を対象に、最適基礎療法注1)単独と比較して本剤400mg 1日2回投与と最適基礎療法を併用した場合の安全性及び抗HIV活性を評価した。無作為化では、プロテアーゼ阻害剤に対する薬剤耐性の程度(1剤又は2剤以上)及び最適基礎療法でのenfuvirtide使用の有無によって層別化した。最適基礎療法は、無作為化前に、薬剤耐性検査(遺伝子型解析/表現型解析)及び抗HIV治療歴に基づいて治験責任医師が選択した。
本剤400mg 1日2回投与群とプラセボ群における患者背景を表1に示す。
注1) 薬剤耐性検査及び抗HIV治療歴に基づいて治験責任医師が無作為化前に選択した、患者ごとの抗HIV薬の最適組み合せ療法
| BENCHMRK 1及び2の |
本剤400mg 1日2回+ 最適基礎療法 (N=462) |
プラセボ+ 最適基礎療法 (N=237) |
|---|---|---|
| 性別 n(%) | ||
| 男性 | 405(87.7) | 210(88.6) |
| 女性 | 57(12.3) | 27(11.4) |
| 人種 n(%) | ||
| 白人 | 301(65.2) | 173(73.0) |
| 黒人 | 65(14.1) | 26(11.0) |
| アジア系 | 16(3.5) | 6(2.5) |
| ヒスパニック | 53(11.5) | 19(8.0) |
| その他 | 27(5.8) | 13(5.5) |
| 年齢(歳) | ||
| 中央値(最小値、最大値) | 45.0(16~74) | 45.0(17~70) |
| CD4リンパ球数 | ||
| 中央値(最小値、最大値)、cells/mm3 | 119(1~792) | 123(0~759) |
| ≦50cells/mm3、n(%) | 146(31.6) | 78(32.9) |
| 50<~≦200cells/mm3、n(%) | 173(37.4) | 85(35.9) |
| 血漿中HIV RNA量 | ||
| 中央値(最小値、最大値)、log10 copies/mL | 4.8(2.3~5.9) | 4.7(2.3~5.9) |
| >100,000copies/mL、n(%) | 165(35.7) | 78(32.9) |
| AIDS確定診断 n(%) | ||
| あり | 427(92.4) | 215(90.7) |
| 抗HIV薬の使用歴、中央値(第1四分位点、第3四分位点) | ||
| 抗HIV薬の使用年数 | 10.1(7.3~12.1) | 10.2(7.9~12.4) |
| 使用抗HIV薬の種類数 | 12.0(9~15) | 12.0(9~14) |
| 肝炎ウイルスの重複感染注2)n(%) | ||
| B型又はC型肝炎の感染なし | 385(83.3) | 200(84.4) |
| B型肝炎の感染のみ | 36(7.8) | 7(3.0) |
| C型肝炎の感染のみ | 37(8.0) | 28(11.8) |
| B型及びC型肝炎の重複感染 | 4(0.9) | 2(0.8) |
| 層別 n(%) | ||
| 最適基礎療法にenfuvirtideを含む | 175(37.9) | 89(37.6) |
| 2剤以上のプロテアーゼ阻害剤に耐性 | 447(96.8) | 226(95.4) |
| 注2) B型肝炎ウイルス表面抗原陽性又はC型肝炎ウイルス抗体陽性 | ||
本剤400mg 1日2回投与群及びプラセボ群におけるベースライン時の最適基礎療法の背景因子の比較を表2に示す。
| BENCHMRK 1及び2の |
本剤400mg 1日2回+ 最適基礎療法 (N=462) |
プラセボ+ 最適基礎療法 (N=237) |
|---|---|---|
| 最適基礎療法中に含まれる抗HIV薬の数 | ||
| 中央値(最小値、最大値) | 4.0(1~7) | 4.0(2~7) |
| 表現型耐性検査において感受性を示した最適基礎療法中のプロテアーゼ阻害剤の数注3) | ||
| 0 | 165(35.7) | 96(40.5) |
| 1以上 | 278(60.2) | 137(57.8) |
| 表現型感受性スコア(PSS)‡ | ||
| 0 | 67(14.5) | 43(18.1) |
| 1 | 144(31.2) | 71(30.0) |
| 2 | 142(30.7) | 66(27.8) |
| 3以上 | 85(18.4) | 48(20.3) |
| 遺伝子型感受性スコア(GSS)注4) | ||
| 0 | 116(25.1) | 65(27.4) |
| 1 | 177(38.3) | 95(40.1) |
| 2 | 111(24.0) | 49(20.7) |
| 3以上 | 51(11.0) | 23(9.7) |
|
||
BENCHMRK 1及び2試験において、無作為化され、本剤400mg 1日2回(本剤の推奨用量)又は対照薬の投与を受けた全患者699例の48及び96週時の転帰を表3に示す。
| BENCHMRK 1及び2の |
48週時 | 96週時 | ||
|---|---|---|---|---|
| 本剤 400mg 1日2回+ 最適基礎療法 (N=462) n(%) |
プラセボ+ 最適基礎療法 (N=237) n(%) |
本剤 400mg 1日2回+ 最適基礎療法 (N=462) n(%) |
プラセボ+ 最適基礎療法 (N=237) n(%) |
|
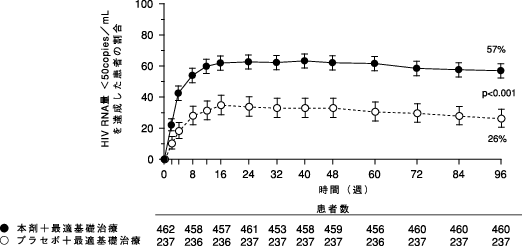
| HIV RNA量 <400copies/mLの患者注5) |
332 (72.3) |
88 (37.1) |
283 (61.5) |
67 (28.3) |
| HIV RNA量 <50copies/mLの患者注5) |
285 (62.1) |
78 (32.9) |
262 (57.0) |
62 (26.2) |
| HIV RNA量 >1Log10 copies/mLの低下(drop)又はHIV RNA量<400copies/mLの患者注5) |
348 (75.8) |
94 (39.7) |
294 (63.9) |
69 (29.1) |
| HIV RNA量のベースラインからの平均変化量(Log10 copies/mL)注5) | -1.71 | -0.78 | -1.51 | -0.60 |
| CD4リンパ球数のベースラインからの平均変化量(cells/mm3)注5) | 109.4 | 44.6 | 123.4 | 48.9 |
| ウイルス学的失敗(確証)注6) | 105 (22.7) |
134 (56.5) |
150 (32.5) |
148 (62.4) |
| ノンレスポンダー | 12 (2.6) |
72 (30.4) |
12 (2.6) |
72 (30.4) |
| 再上昇 | 93 (20.1) |
62 (26.2) |
138 (29.9) |
76 (32.1) |
| 死亡注7) | 10 (2.2) |
6 (2.5) |
13 (2.8) |
6 (2.5) |
| AIDS指標疾患注7) | 17 (3.7) |
11 (4.6) |
18 (3.9) |
11 (4.6) |
| 有害事象(臨床症状)による中止注7) | 10 (2.2) |
7 (3.0) |
16 (3.5) |
10 (4.2) |
| 有害事象(臨床検査値)による中止注7) | 1 (0.2) |
0 (0.0) |
1 (0.2) |
0 (0.0) |
| 他の理由による中止注7)注8) | 11 (2.4) |
4 (1.7) |
38 (8.2) |
19 (8.0) |
|
||||
HIV RNA量<50copies/mLを達成した患者の割合の推移(95%信頼区間)を図1に示す(未完了例=失敗例;NC=F法)。
ベースラインにおける表現型及び遺伝子型感受性スコア別のウイルス学的治療効果(96週時)を表4に示す。
| BENCHMRK 1及び2の |
本剤400mg 1日2回+ 最適基礎療法(N=462) |
プラセボ+最適基礎療法 (N=237) |
||||
|---|---|---|---|---|---|---|
| n | HIV RNA量 <400copies/mL の患者割合(%) |
HIV RNA量 <50copies/mL の患者割合(%) |
n | HIV RNA量 <400copies/mL の患者割合(%) |
HIV RNA量 <50copies/mL の患者割合(%) |
|
| 表現型感受性スコア(PSS)注10) | ||||||
| 0 | 63 | 51 | 48 | 43 | 5 | 5 |
| 1 | 131 | 69 | 65 | 68 | 26 | 24 |
| 2 | 134 | 74 | 69 | 60 | 37 | 35 |
| 3以上 | 74 | 62 | 54 | 40 | 53 | 48 |
| 遺伝子型感受性スコア(GSS)注10) | ||||||
| 0 | 111 | 46 | 41 | 64 | 5 | 5 |
| 1 | 160 | 76 | 72 | 89 | 31 | 28 |
| 2 | 102 | 75 | 70 | 41 | 61 | 61 |
| 3以上 | 45 | 62 | 53 | 21 | 48 | 38 |
|
||||||
17.1.2 抗HIV治療未経験患者を対象にした海外第III相臨床試験(STARTMRK)
STARTMRK(無作為化、二重盲検、実薬対照試験)は、HIV RNA量>5,000copies/mLの治療未経験HIV感染患者を対象に、EFV+FTC+TDFに対する本剤400mg 1日2回投与+FTC+TDFの安全性及び抗HIV活性を評価する試験であり、無作為化にあたっては、スクリーニング時のHIV RNA量(≦50,000copies/mL又は>50,000copies/mL)及び肝炎ウイルスの重複感染状況によって層別割付を実施した。
本剤400mg 1日2回投与群とEFV投与群における患者背景を表5に示す。
| 本剤400mg 1日2回 (N=281) |
EFV 600mg 就寝時 (N=282) |
|
|---|---|---|
| 性別 n(%) | ||
| 男性 | 227(80.8) | 231(81.9) |
| 女性 | 54(19.2) | 51(18.1) |
| 人種 n(%) | ||
| 白人 | 116(41.3) | 123(43.6) |
| 黒人 | 33(11.7) | 23(8.2) |
| アジア系 | 36(12.8) | 32(11.3) |
| ヒスパニック系 | 60(21.4) | 67(23.8) |
| アメリカ原住民 | 1(0.4) | 1(0.4) |
| 混血 | 35(12.5) | 36(12.8) |
| 地域 n(%) | ||
| ラテンアメリカ | 99(35.2) | 97(34.4) |
| 東南アジア | 34(12.1) | 29(10.3) |
| 北アメリカ | 82(29.2) | 90(31.9) |
| EU/オーストラリア | 66(23.5) | 66(23.4) |
| 年齢(歳) | ||
| 18~64、n(%) | 279(99.3) | 278(98.6) |
| ≧65、n(%) | 2(0.7) | 4(1.4) |
| 平均(標準偏差) | 37.6(9.0) | 36.9(10.0) |
| 中央値(最小値、最大値) | 37.0(19~67) | 36.0(19~71) |
| CD4リンパ球数(cells/mm3) | ||
| N注11) | 281 | 281 |
| 平均(標準偏差) | 218.9(124.2) | 217.4(133.6) |
| 中央値(最小値、最大値) | 212.0(1~620) | 204.0(4~807) |
| 血漿中HIV RNA量(log10 copies/mL) | ||
| N† | 281 | 282 |
| 平均(標準偏差) | 5.0(0.6) | 5.0(0.6) |
| 中央値(最小値、最大値) | 5.1(2.6~5.9) | 5.0(3.6~5.9) |
| 血漿中HIV RNA量(copies/mL) | ||
| N注11) | 281 | 282 |
| 幾何平均 | 103,205 | 106,215 |
| 中央値(最小値、最大値) | 114,000(400~750,000) | 104,000(4,410~750,000) |
| AIDS確定診断 n(%) | ||
| あり | 52(18.5) | 59(20.9) |
| 層別 n(%) | ||
| スクリーニング時HIV RNA量≦50,000copies/mL | 75(26.7) | 80(28.4) |
| B型又はC型肝炎陽性注12) | 18(6.4) | 16(5.7) |
| HIVサブタイプ n(%) | ||
| サブタイプB | 219(77.9) | 230(81.6) |
| サブタイプB以外注13) | 59(21.0) | 47(16.7) |
| 不明 | 3(1.1) | 5(1.8) |
| ベースライン血漿中HIV RNA量注11)n(%) | ||
| ≦50,000copies/mL | 79(28.1) | 84(29.8) |
| >50,000copies/mL | 202(71.9) | 198(70.2) |
| ≦100,000copies/mL | 127(45.2) | 139(49.3) |
| >100,000copies/mL | 154(54.8) | 143(50.7) |
| ベースラインのCD4リンパ球数 n(%) | ||
| ≦50cells/mm3 | 27(9.6) | 31(11.0) |
| >50cells/mm3かつ≦200cells/mm3 | 104(37.0) | 105(37.2) |
| >200cells/mm3 | 150(53.4) | 145(51.4) |
| 不明 | 0(0.0) | 1(0.4) |
|
||
STARTMRKにおける48及び240週時の転帰、ウイルス学的抑制及び免疫学的効果の解析結果における群間差及び95%信頼区間を表6に示す。
| STARTMRK (無作為化試験) |
48週時 | 240週時 | ||||
|---|---|---|---|---|---|---|
| 本剤 400mg 1日2回 (N=281) n(%) |
EFV 600mg 就寝時 (N=282) n(%) |
差 (本剤–EFV) (信頼区間注15)) |
本剤 400mg 1日2回 (N=281) n(%) |
EFV 600mg 就寝時 (N=282) n(%) |
差 (本剤–EFV) (信頼区間注15)) |
|
| HIV RNA量<50copies/mLの患者注15) | 241 (86.1) |
230 (81.9) |
4.2% (-1.9, 10.3) |
198 (71.0) |
171 (61.3) |
9.5% (1.7, 17.3) |
| HIV RNA量<400copies/mLの患者注15) | 252 (90.0) |
241 (85.8) |
4.1% (-1.3, 9.7) |
206 (73.8) |
181 (64.9) |
8.8% (1.2, 16.4) |
| CD4リンパ球数のベースラインからの平均変化量(cells/mm3)注15) | 189.1 | 163.3 | 25.8 (4.4, 47.2) |
373.7 | 311.6 | 62.1 (21.9, 102.2) |
| ウイルス学的失敗(確証)注16) | 27 (9.6) |
39 (13.8) |
55 (19.6) |
59 (20.9) |
||
| ノンレスポンダー | 10 (3.6) |
24 (8.5) |
10 (3.6) |
24 (8.5) |
||
| 再上昇 | 17 (6.0) |
15 (5.3) |
45 (16.0) |
35 (12.4) |
||
| 死亡 | 2 (0.7) |
0 (0.0) |
5 (1.8) |
5 (1.8) |
||
| 有害事象(臨床症状)による中止 | 8 (2.8) |
17 (6.0) |
14 (5.0) |
25 (8.9) |
||
| 有害事象(臨床検査値)による中止 | 0 (0.0) |
1 (0.4) |
0 (0.0) |
3 (1.1) |
||
| 他の理由による中止注17) | 12 (4.3) |
15 (5.3) |
51 (18.1) |
60 (21.3) |
||
|
||||||
血漿中HIV RNA量<50copies/mLを達成した患者割合の推移を投与群別に図2に示す。本剤投与群はEFV投与群より早期にウイルス学的抑制(HIV RNA量<50copies/mL)を達成した(両投与群とも、FTC+TDFを併用)。240週時には、本剤400mg 1日2回投与群の71%が、及びEFV群の61%がHIV RNA量<50copies/mLに達した(未完了例=失敗例;NC=F法)。
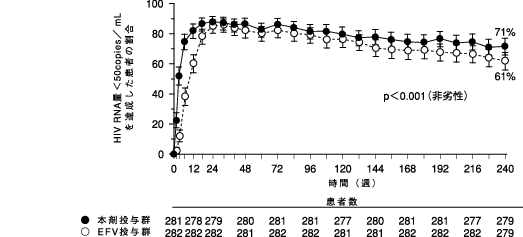
抗HIV治療未経験患者を対象にした複数の抗HIV薬による治療を行うSTARTMRK試験では、以下の背景因子及び予後因子のベースライン値にかかわらず、本剤+FTC+TDFの投与はEFV+FTC+TDFの投与に比較して、一貫して持続的なウイルス学的抑制及び免疫学的効果を示した。:ベースライン血漿中HIV RNA量、ベースラインのCD4リンパ球数、患者背景(年齢、性別、地域、人種を含む)、ウイルス性肝炎(B型/C型肝炎)の重複感染の状態及びHIVサブタイプ(サブタイプB以外とサブタイプBグループとを比較)
本剤の持続的効果はすべてのHIVサブタイプにおいて認められ、サブタイプB及びサブタイプB以外の患者ではそれぞれ89.6%(155/173例)及び87.0%(40/46例)が、240週時にHIV RNA量<50copies/mLに達した(Observed Failure法)。
240週間の治療を通して、本剤の血清脂質に対する影響は軽微であり、総コレステロール、HDLコレステロール、LDLコレステロール、トリグリセリド及び非HDLコレステロールはわずかに増加した。EFV投与群では、総コレステロール、HDLコレステロール、LDLコレステロール、トリグリセリド及び非HDLコレステロールのベースラインからの平均変化量は本剤投与群と比較してEFV投与群で有意に大きかった。
STARTMRKにおいて、本剤(400mg 1日2回)とエムトリシタビン(FTC)及びテノホビルジソプロキシルフマル酸塩(TDF)の併用投与群(281例)の2%以上に認められた中等度又は重度の副作用は、悪心(2.8%)、頭痛(3.9%)、不眠症(3.6%)であった。
18. 薬効薬理
18.1 作用機序
HIVインテグラーゼは、HIV遺伝子にコードされたウイルス複製に必要な酵素であり、ラルテグラビルは、HIVインテグラーゼの触媒活性を阻害する。HIVインテグラーゼの阻害により、HIV感染初期において、HIVゲノムの宿主細胞ゲノムへの共有結合的挿入又は組込みが阻害される。組み込まれなかったHIVゲノムは、感染性ウイルス粒子を新たに産生することができないため、ウイルスの感染拡大が阻止される。なお、ラルテグラビルは、DNAポリメラーゼα、β、γを含むヒトホスホリルトランスフェラーゼに対し、顕著な阻害作用を示さなかった。
18.2 抗ウイルス作用(in vitro)
ヒトTリンパ球系細胞に、その細胞に適応したHIV-1変異株H9IIIBを感染させた試験系において、ウイルス増殖に対するラルテグラビルの95%阻害濃度(IC95)は31±20nMであった(無処置感染細胞との比較)。また、マイトジェン活性化ヒト末梢血単核細胞に、5種のサブタイプB以外からの分離株や逆転写酵素阻害剤及びプロテアーゼ阻害剤耐性分離株を含む様々なHIV-1初代臨床分離株を感染させた試験系において、ウイルス増殖に対するラルテグラビルのIC95値は、6~50nMであった。また、singlecycle infection assayにおいて、ラルテグラビルは5種のサブタイプB以外及び5種の組換え型など23種のHIV分離株の感染を5~12nMのIC50値で阻害した。さらに、ラルテグラビルは、CEMx174細胞においてHIV-2分離株の複製を阻害した(IC95=6nM)。HIV-1変異株H9ⅢBを感染させたヒトTリンパ球系細胞に対して、ラルテグラビルと核酸系逆転写酵素阻害剤(ジドブジン、ザルシタビン、サニルブジン、アバカビル、テノホビル、ジダノシン又はラミブジン)、非核酸系逆転写酵素阻害剤(エファビレンツ、ネビラピン又はデラビルジン)、プロテアーゼ阻害剤(インジナビル、サキナビル、リトナビル、アンプレナビル、ロピナビル、ネルフィナビル又はアタザナビル)又は融合阻害剤(enfuvirtide)とを併用したところ、相加的若しくは相乗的な抗HIV活性が認められた。
18.3 薬剤耐性
In vitro試験又はラルテグラビル投与患者でみられた、ラルテグラビル耐性を示すHIV-1インテグラーゼの変異は、概して、143番目のチロシン(Y)のシステイン(C)、ヒスチジン(H)又はアルギニン(R)への置換、148番目のグルタミン(Q)のヒスチジン(H)、リシン(K)又はアルギニン(R)への置換、あるいは155番目のアスパラギン(N)のヒスチジン(H)への置換に、さらに1つ以上の変異(L74I/M、E92Q、E138A/K、G140A/S又はV151I等)が加わるものであった。
単一の一次変異(Q148H/K/R、あるいはN155H)を含む組換えウイルスでは、in vitroにおいて、ラルテグラビル感受性の低下及び複製能力の低下がみられた。また二次的な変異では、ラルテグラビル感受性のさらなる低下と、複製能力の代償的ウイルス変異がときにみられた。
19. 有効成分に関する理化学的知見
| 一般的名称 | ラルテグラビルカリウム(raltegravir potassium) |
|---|---|
| 化学名 | Monopotassium 4-[(4-fluorobenzyl)carbamoyl]-1-methyl-2-(1-methyl-1-{[(5-methyl-1,3,4-oxadiazol-2-yl)carbonyl]amino}ethyl)-6-oxo-1,6-dihydropyrimidin-5-olate |
| 分子式 | C20H20FKN6O5 |
| 分子量 | 482.51 |
| 性状 | 白色~帯灰白色の粉末。水にやや溶けやすく、メタノールに溶けにくく、エタノール又はアセトニトリルに極めて溶けにくく、2-プロパノールにほとんど溶けない。 |
| 化学構造式 | 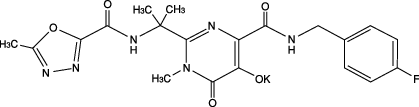 |
22. 包装
60錠[瓶、バラ、乾燥剤入り]
23. 主要文献
- 社内資料:胎盤通過に関する検討(2008年6月24日承認、CTD 2.6.4.4)
- 社内資料:乳汁移行に関する検討(2008年6月24日承認、CTD 2.6.4.6)
- 厚生労働省:重篤副作用疾患別対応マニュアル 薬剤性過敏症症候群
- 社内資料:単回及び反復投与に関する検討(2008年6月24日承認、CTD 2.7.2.2、CTD 2.7.2.3)
- Markowitz, M. et al. J Acquir Immune Defic Syndr. 2006;43(5):509-15.
- Brainard, DM. et al. J Clin Pharmacol. 2011;51(3):422-7.
- Croteau, D. et al. Antimicrob Agents Chemother. 2010;54(12):5156-60.
- Yilmaz, A. et al. PLoS One. 2009;4(9):e6877.
- Kassahun, K. et al. Drug Metab Dispos. 2007;35(9):1657-63.
- 社内資料:腎機能障害による薬物動態への影響に関する検討(2008年6月24日承認、CTD 2.7.2.3)
- 社内資料:肝機能障害による薬物動態への影響に関する検討(2008年6月24日承認、CTD 2.7.2.3)
- Wenning, LA. et al. Clin Pharmacol Ther. 2009;85(6):623-7.
- 社内資料:代謝酵素及びトランスポーターに対する阻害及び誘導作用に関する検討(2008年6月24日承認、CTD 2.6.4.5)
- Iwamoto, M. et al. J Clin Pharmacol. 2008;48(2):209-14.
- Iwamoto, M. et al. Clin Infect Dis. 2008;47(1):137-40.
- Iwamoto, M. et al. Clin Infect Dis. 2009;48(4):489-92.
- Anderson, MS. et al. Br J Clin Pharmacol. 2011;71(4):616-20.
- Anderson, MS. et al. J Clin Pharmacol. 2010;50(12):1461-6.
- Wenning, LA. et al. Antimicrob Agents Chemother. 2008;52(9):3253-8.
- Markowitz, M. et al. J Acquir Immune Defic Syndr. 2007;46(2):125-33.
- Anderson, MS. et al. Antimicrob Agents Chemother. 2008;52(12):4228-32.
- Iwamoto, M. et al. Antimicrob Agents Chemother. 2008;52(12):4338-43.
- Hanley, WD. et al. Antimicrob Agents Chemother. 2009;53(7):2752-5.
- Wenning, LA. et al. Antimicrob Agents Chemother. 2009;53(7):2852-6.
24. 文献請求先及び問い合わせ先
MSD株式会社 MSDカスタマーサポートセンター
東京都千代田区九段北1-13-12
医療関係者の方:フリーダイヤル 0120-024-961
26. 製造販売業者等
26.1 製造販売元
MSD株式会社
東京都千代田区九段北1-13-12