ビラミューン錠200の添付文書
- 商品名:
- ビラミューン錠200
- 一般名:
- ネビラピン
- 略称 :
- NVP

添付文書の読み方
ここで提供している添付文書情報は、2025年3月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
抗ウイルス化学療法剤
ネビラピン
- *2023年8月改訂(第2版)
- 2021年7月改訂(第1版)
- 劇薬、処方箋医薬品注)
注)注意-医師等の処方箋により使用すること - ®=登録商標
| 日本標準商品分類番号 | 87625 |
|---|
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 3年 |
| 承認番号 | 21000AMY00263000 |
|---|---|
| 販売開始 | 1998年12月 |
1. 警告
1.1 皮膚障害
本剤の投与により、中毒性表皮壊死症(Lyell症候群)、皮膚粘膜眼症候群(Stevens-Johnson症候群)、過敏症症候群を含め、重篤で致死的な皮膚障害が発現することがあるので、次の事項に注意すること。
- 本剤による発疹は、投与開始後概ね18週までに(重篤な発疹は投与開始後概ね6週までに)発現する場合が多いので、当該期間中は特に観察を十分に行うこと。
- 重篤な発疹、又は以下の症状を伴う発疹が発現した場合には、本剤の投与を中止すること。
発熱、水疱、口内病変、結膜炎、顔面や四肢等の腫脹、筋肉痛、関節痛、又は全身倦怠感
なお、必要に応じ、専門医を受診させるなど適切な処置を行うこと。 - 投与中止後も症状が増悪するおそれがあるので、患者の状態を十分観察すること。
- 本剤の投与により重篤な発疹、又は全身症状を伴う発疹が発現した患者には、再投与しないこと。[2.2、8.1、11.1.1参照]
1.2 肝機能障害
本剤の投与により、肝不全などの重篤で致死的な肝機能障害が発現することがあるので、次の事項に注意すること。
- 投与開始に際しては肝機能検査を含む臨床検査を実施し、更に投与開始後6カ月間は少なくとも1カ月に1回、定期的かつ必要に応じて肝機能検査を行うなど、患者の状態を十分に観察すること。
- 異常が認められた場合(γ-GTPを除く)には、投与を中止するなど適切な処置を行うこと。
- 投与中止後も症状が増悪するおそれがあるので、患者の状態を十分観察すること。
- 本剤の投与により肝機能障害が発現した患者には再投与しないこと。[2.4、8.1、8.4、11.1.3参照]
2. 禁忌(次の患者には投与しないこと)
2.1
本剤の成分に対し過敏症の既往歴のある患者
2.2
本剤の投与により重篤な発疹、又は全身症状を伴う発疹が発現した患者[1.1、8.1、11.1.1参照]
2.3
重篤な肝機能障害のある患者[9.3.1参照]
2.4
本剤の投与により肝機能障害が発現した患者[1.2、8.1、8.4、11.1.3参照]
2.5
ケトコナゾール(経口剤:国内未発売)を投与中の患者[10.1参照]
2.6
経口避妊薬を投与中の患者(避妊を目的とするホルモン療法も含む)[8.1、10.1参照]
3. 組成及び性状
3.1 組成
| 販売名 | ビラミューン錠200 |
|---|---|
| 有効成分 | 1錠中ネビラピン 200mg |
| 添加剤 | 結晶セルロース、乳糖水和物、ポビドンK25、デンプングリコール酸ナトリウム、軽質無水ケイ酸、ステアリン酸マグネシウム |
3.2 性状
| 販売名 | ビラミューン錠200 |
|---|---|
| 剤形 | 白色の素錠(割線) |
| 外形 |  |
| 長径 | 19.1mm |
| 短径 | 9.3mm |
| 厚さ | 6.5mm |
| 重さ | 800mg |
| 識別コード |  54 193 54 193 |
4. 効能又は効果
HIV-1感染症
5. 効能又は効果に関連する注意
5.1
無症候性HIV感染症に関する治療開始の指標はCD4リンパ球数500/mm3以下もしくはHIV RNA量5000copies/mL(RT-PCR法)以上との国際的な勧告がある。従って、本剤の使用にあたってはCD4リンパ球数及びHIV RNA量を確認すること。
6. 用法及び用量
通常、成人にはネビラピンとして1回200mgを1日1回、14日間経口投与する。その後、維持量として1日400mgを2回に分割して経口投与する。なお、年齢、症状により適宜増減する。投与に際しては必ず他の抗HIV薬と併用すること。
7. 用法及び用量に関連する注意
7.1
本剤は少なくとも1種類の抗レトロウイルス剤(ヌクレオシド系逆転写酵素阻害剤又はHIVプロテアーゼ阻害剤)と必ず併用投与し、単独投与しないこと。
単独投与すると、いずれの症例においても本剤に耐性を示すウイルスが急速に出現することが報告されている。[8.2参照]
7.2
ヒト免疫不全ウイルス(HIV)は感染初期から多種多様な変異株を生じ、薬剤耐性を発現しやすいことが知られているので、本剤は他の抗HIV薬と併用すること。
7.3
非ヌクレオシド系逆転写酵素阻害剤(NNRTI)を2剤併用したときの有用性が示されていない。他のNNRTIとの併用は避けることが望ましい。
7.4
HIV治療に対して種々の国際的なガイドラインが出されており、現時点では、非ヌクレオシド系逆転写酵素阻害剤である本剤でHIV治療を行う際には、2種類のヌクレオシド系逆転写酵素阻害剤との併用が推奨されている。ヌクレオシド系逆転写酵素阻害剤との2剤併用(ネビラピン+ジドブジン)より、3剤併用(ネビラピン+ジドブジン+ジダノシン)で優れた臨床効果が得られている。
7.5
ネビラピンは他の非ヌクレオシド系逆転写酵素阻害剤と交叉耐性を示すことがある。非ヌクレオシド系逆転写酵素阻害剤に対して交叉耐性を示すHIVウイルス株が認められたとの報告がある。
7.6
本剤の投与は1日200mgより開始し、1日400mgの維持量に増量するが、発疹が発現した場合には、発疹が完治するまで本剤の投与量を増量しないこと。
7.7
7日間以上本剤を中止した患者に対して投与を再開する場合には、導入期の用法・用量から始めること。
8. 重要な基本的注意
* 8.1
本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又は患者に代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。
- 本剤はHIV-1感染症の根治療法薬ではないことから、日和見感染を含む感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。
- 本剤の主な副作用は発疹であること。本剤による発疹は投与開始後概ね18週までに(重篤な発疹は投与開始後概ね6週までに)発現しているので、当該期間中は特に注意すること。また、発疹が発現した場合には、直ちに担当医に報告すること。[1.1、2.2、11.1.1参照]
- 本剤の投与により、肝不全などの重篤な肝機能障害の発現が報告されていること。[1.2、2.4、8.4、11.1.3参照]
- 本剤を処方どおり毎日服用すること。また、医師の指示なしに用量を変更しないこと。さらに、服用し忘れた場合には、気づいたときにすぐに服用し、決して次回服用時に2回量を服用しないこと。
- 本剤は他の薬剤と相互作用を示す可能性があるので、他の薬剤の服用の有無について担当医に報告すること。
- 本剤の服用中は経口避妊薬又は他のホルモン療法を避妊目的に使用しないこと。[2.6、10.1参照]
8.2
抗レトロウイルス療法により得られる便益の持続時間は限られているので、本剤による治療中に疾患の進展が認められた場合には、他の抗レトロウイルス療法への変更を考慮すること。[7.1参照]
8.3
CD4値、血漿中HIV-1 RNAコピー数の測定、治療開始時の抗レトロウイルス剤による治療経験の有無の確認を行うとともに肝機能検査を合わせて行うこと。[9.1.1参照]
8.4
肝機能障害があらわれることがあるので、定期的、かつ必要に応じて検査を行うなど患者の状態を十分に観察すること。[1.2、2.4、8.1、11.1.3参照]
8.5
発疹の副作用の発現に伴って肝機能障害の副作用が発現する症例が報告されているので、発疹があらわれた患者では肝機能検査も合わせて行うこと。[9.1.2参照]
8.6
本剤を含む抗HIV薬の多剤併用療法を行った患者で、免疫再構築症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 CD4値が高く(女性:250/mm3以上、男性:400/mm3以上)、血漿中にHIV-1 RNAが検出される(概ね50copies/mL以上)患者
あるいは抗レトロウイルス剤による治療経験がない患者CD4値が低い患者に比べて本剤による肝機能障害の発現率が高い。[8.3参照]
9.1.2 女性の患者
発疹や肝機能障害の発現に十分注意すること。女性の患者では、本剤による発疹や発疹に伴う肝機能障害の発現率が高い。[8.5参照]
9.2 腎機能障害患者
腎障害又はその既往歴のある患者では、本剤の血中濃度に影響を与えるおそれがある。[16.6.1参照]
9.3 肝機能障害患者
9.3.1 重篤な肝機能障害のある患者
投与しないこと。Child-Pugh分類スコア8で中等度から高度の腹水を伴う患者では、肝機能の悪化により本剤の血中濃度増加を招く。[2.3、16.6.2参照]
9.3.2 肝機能障害(重篤な肝機能障害を除く)又はその既往歴のある患者
肝機能障害を増悪させることがある。また、本剤の血中濃度に影響を与えるおそれがある。[16.6.2参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験(ラット、ウサギ)において、以下のことが報告されている。
- ラットの受胎能及び一般生殖能試験において、50mg/kg以上で交尾率、妊娠率、着床数及び生存胎児数の低下、着床前死亡率及び吸収胚率の上昇、新生児数減少と生後体重の増加抑制がみられた。
- ラット及びウサギの胎児器官形成期投与試験において、催奇形性は認められなかったが、高用量群(50mg/kg及び300mg/kg)で母動物及び胎児に体重低下や生存胎児数の減少がみられた。
- ラットの周産期及び授乳期投与試験では、100mg/kgで母動物は18匹中16匹が死亡した(一般状態の悪化による切迫屠殺を含む)。40mg/kgでは次世代児の体重、4日生存率、離乳率の低下がみられ、その生殖能にも影響が認められた。
9.6 授乳婦
授乳を避けさせること。母乳中へ移行することが認められている。
9.7 小児等
小児等を対象とした臨床試験は実施していない。[16.6.3参照]
9.8 高齢者
一般に生理機能(肝機能、腎機能)が低下している。[16.6.4参照]
10. 相互作用
本剤は主として薬物代謝酵素CYP3Aで代謝される。[16.4参照]
10.1 併用禁忌(併用しないこと)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| ケトコナゾール(経口剤:国内未発売) [2.5参照] |
併用によりケトコナゾールの血中濃度が低下し(AUCの低下:63%、Cmaxの低下:40%)、また本剤の血中濃度が上昇(15~28%)したとの報告がある。 | 本剤はCYP3Aを誘導し、また代謝される(自己誘導)が、ケトコナゾールは当該酵素の阻害剤である。 |
| 経口避妊薬(避妊を目的とするホルモン療法も含む) ノルエチステロン・エチニルエストラジオール(シンフェーズ) [2.6、8.1参照] |
本剤が経口避妊薬の血中濃度を低下させることがある。(併用により、エチニルエストラジオールのAUCが20%、Cmaxが6%それぞれ低下、また、ノルエチステロンのAUCが19%、Cmaxが16%それぞれ低下したとの報告がある。) | 機序不明 |
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| HIVプロテアーゼ阻害剤 インジナビル サキナビル リトナビル ホスアンプレナビル |
これらの薬剤の血中濃度が低下した(AUCの低下:インジナビル28%、サキナビル24%、リトナビル10%、Cmaxの低下:インジナビル11%、サキナビル28%、リトナビル10%、Cminの低下:インジナビル38%、リトナビル9%)との報告があるので、患者の状態を十分に観察するなど注意すること。また、本剤200mg1日2回とホスアンプレナビル1400mg1日2回を併用した場合、ホスアンプレナビルについては、活性代謝物であるアンプレナビルの血中濃度が低下し(AUC33%、Cmax25%、Cmin35%)、本剤の血中濃度が上昇した(AUC29%、Cmax25%、Cmin34%)との報告があるので、患者の状態を十分に観察するなど注意すること。 | 本剤はCYP3Aを誘導し、また代謝される(自己誘導)が、これらの薬剤は当該酵素により代謝される。 |
| CYP3A酵素阻害剤 シメチジン マクロライド系抗生物質 アジスロマイシン等 イトラコナゾール |
本剤の定常状態におけるCminが上昇したとの報告(シメチジンとの併用:7%、マクロライド系抗生物質との併用:12%、イトラコナゾールとの併用:17%)があるので、併用の開始、用量の変更並びに中止時には、副作用の発現に注意し、患者の状態を十分に観察するなど注意すること。 | 本剤はCYP3Aを誘導し、また代謝される(自己誘導)が、シメチジン、マクロライド系抗生物質及びイトラコナゾールは当該酵素の阻害剤であり、リファンピシン等は当該酵素の誘導剤である。 また、セイヨウオトギリソウに含有される成分が、当該酵素を誘導するとの報告がある。 上記以外にも、併用薬剤がCYP3Aで代謝を受ける薬剤である場合には相互に影響を受ける可能性が考えられる。 |
| CYP3A酵素誘導剤 リファンピシン リファブチン |
リファンピシンとの併用において定常状態における本剤のAUCが58%、Cmaxが50%、Cminが68%低下したとの報告がある。 またリファブチンとの併用において有意ではないが定常状態におけるリファブチンのAUCが12%増加し、Cminは3%低下し、Cmaxは有意に20%増加したとの報告がある。リファブチンの活性代謝物濃度に変化は見られなかった。また、本剤の全身クリアランスが9%増加した。 以上のことから併用の開始、用量の変更並びに中止時には、副作用の発現に注意し、患者の状態を十分に観察するなど注意すること。 |
|
| セイヨウオトギリソウ(St.John’s Wort,セント・ジョーンズ・ワート)含有食品 | 本剤の代謝が促進され血中濃度が低下するおそれがあるので、本剤投与時はセイヨウオトギリソウ含有食品を摂取しないよう注意すること。 | |
| 他の(上記以外の)CYP3A酵素で代謝を受ける薬剤 トリアゾラム |
併用薬剤の血中濃度又は本剤の血中濃度が変動するおそれがあるので、患者の状態を十分に観察するなど注意すること。 | |
| ワルファリン | 血液凝固時間が変化することがあるので、プロトロンビン時間の変化に十分注意すること。 | 本剤はCYP3Aによるワルファリン(R-ワルファリン)の代謝に影響を与える可能性が考えられる。 |
| 避妊が目的でないホルモン療法(経口避妊薬を含む) エストラジオール |
本剤が併用薬剤の血中濃度を低下させることがあるので、ホルモン療法の治療効果を確認すること。 | 機序不明 |
11. 副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1 重大な副作用
11.1.1 中毒性表皮壊死症(Lyell症候群)、皮膚粘膜眼症候群(Stevens-Johnson症候群)(0.7%)
これらの重篤な発疹は本剤投与開始後概ね6週までに発現する場合が多いので、この期間は特に観察を十分に行い、重篤な発疹、又は以下の症状を伴う発疹が発現した場合には、本剤の投与を中止すること。また、このような患者には再投与しないこと。
発熱、水疱、口内病変、結膜炎、顔面や四肢等の腫脹、筋肉痛、関節痛、又は全身倦怠感
なお、必要に応じ、専門医を受診させるなど適切な処置を行うこと。また、投与中止後も症状が増悪するおそれがあるので患者の状態を十分観察すること。[1.1、2.2、8.1参照]
11.1.2 過敏症症候群(頻度不明)
初期症状として発疹、発熱がみられ、さらにリンパ節腫脹、肝機能障害、白血球増加、好酸球増多、異型リンパ球出現等を伴う遅発性の重篤な過敏症状(薬剤性過敏症症候群)があらわれることがある。なお、発疹、発熱、肝機能障害等の症状が再燃あるいは遷延化することがあるので注意すること。
11.1.3 肝炎(劇症肝炎を含む)、肝機能障害(AST、ALT、γ-GTP、Al-P、総ビリルビン等の上昇)、黄疸、肝不全(6.3%)
肝機能検査値の異常が認められた場合は、本剤の投与を中止すること。[1.2、2.4、8.1、8.4参照]
11.1.4 顆粒球減少、うつ病、幻覚、錯乱、脱水症、心筋梗塞、出血性食道潰瘍、全身痙攣、髄膜炎(4.2%)
11.1.5 アナフィラキシー(0.2%)
アナフィラキシー(発疹、じん麻疹、血管浮腫等)があらわれることがある。
11.2 その他の副作用
| 5%以上 | 5%未満 | 頻度不明 | |
|---|---|---|---|
| 循環器 | 潮紅、頻脈、心悸亢進、起立性低血圧、肺塞栓症 | 血圧上昇 | |
| 消化器 | 嘔気(6.2%) | 下痢、嘔吐、消化不良、腹痛、潰瘍性口内炎、食欲不振、鼓腸放屁、血清アミラーゼ上昇、便秘、歯肉炎、唾液増加、嚥下障害、胃潰瘍(出血性)、直腸出血、食欲亢進、膵炎、胃炎、口渇、直腸障害 | |
| 精神神経系 | 傾眠(5.2%)、頭痛(5.3%) | めまい、神経過敏、不眠症、思考異常、激越、緊張亢進、感情不安定、不随意筋収縮、末梢神経障害、知覚減退、運動過多、不安、異夢、偏頭痛、眩暈、昏迷、言語障害、神経障害、多幸症、記憶力低下、感情鈍麻、悪夢、インポテンス、リビドー減退、精神運動発達障害、攻撃性反応 | |
| 皮膚 | 発疹(12.2%) | そう痒、紫斑、皮膚疾患、皮膚剝離、皮膚変色、脱毛、湿疹、紅斑性発疹 | 斑状丘疹性皮疹 |
| 感覚器 | 味覚倒錯、味覚喪失、感覚異常、結膜炎、視力異常、羞明、耳鳴、眼球乾燥 | ||
| 肝臓 | 肝腫、胆嚢炎 | ||
| 腎臓 | 腎機能異常、低リン酸血症、乏尿、尿細管障害 | ||
| 血液 | 貧血、リンパ節症 | 好酸球増加 | |
| 筋・骨格 | CK上昇、筋肉痛、関節痛、筋力低下 | ||
| 呼吸器 | 咳、咽頭炎、上気道炎、呼吸困難 | ||
| その他 | 発熱(6.3%) | 疲労、多汗、悪寒、倦怠感、体重減少、疼痛、顔面浮腫、胸痛、副鼻腔炎、アレルギー反応、背部痛、ほてり、頻尿、インフルエンザ様症候群、虚血性壊死、高トリグリセライド血症、月経異常、膿瘍、尿路感染、寄生虫感染、モニリア症、中耳炎、単純疱疹、鼻出血、鼻炎、人格障害、光線過敏性反応、神経炎 | 体脂肪の再分布/蓄積 |
13. 過量投与
13.1 症状
本剤の過量服用により浮腫、結節性紅斑、疲労、発熱、頭痛、不眠、嘔気、肺浸潤、発疹、眩暈、嘔吐、トランスアミナーゼ値上昇、体重減少の発現が報告されている。
15. その他の注意
15.2 非臨床試験に基づく情報
ラット及びマウスに長期投与したところ、対照群に比較して肝腫瘍の発生が有意に増加したとの試験成績がある。
16. 薬物動態
16.1 血中濃度
健康成人男性に本剤50、100、200、400mgを単回経口投与した場合、Cmax、AUC0-∞は投与量に比例して増加した。なお、半減期は約40時間であった。
| 用量 (mg) |
例数 | Cmax (μg/mL) |
Tmax (hr) |
AUC0-∞ (μg・hr/mL) |
CL/F (mL/hr/kg) |
Vdss/F (L/kg) |
T1/2 (hr) |
|---|---|---|---|---|---|---|---|
| 50 | 6 | 0.7±0.1 | 2.8±1.5 | 33.0±5.6 | 25.1±3.9 | 1.4±0.1 | 39.6±8.0 |
| 100 | 6 | 1.2±0.2 | 2.2±1.0 | 77.1±21.1 | 22.0±5.9 | 1.4±0.2 | 46.9±12.7 |
| 200 | 6 | 2.3±0.3 | 2.2±1.5 | 172.4±37.6 | 19.3±3.3 | 1.3±0.1 | 47.1±7.2 |
| 400 | 6 | 3.9±0.6 | 5.7±5.1 | 282.6±31.8 | 23.1±3.3 | 1.3±0.1 | 39.6±2.2 |
また、HIV感染患者にネビラピン200mg/日を2週間、300mg/日を4週間、400mg/日を4週間の計10週間、経口投与した時の最低薬物血漿中濃度はそれぞれ2.8±0.6μg/mL(n=10)、3.5±0.7μg/mL(n=7)、4.7±1.2μg/mL(n=6)であった。
健康成人男性(n=15)及び女性(n=15)にネビラピン200mgを単回経口投与した時の最高血漿中濃度(約2.0±0.4μg/mL)は、投与後4時間までに得られ、半減期は44時間であった(外国人データ)。
| 性別 | Cmax (μg/mL) |
AUC0-∞ (μg・hr/mL) |
CL/F (mL/hr/kg) |
Vdss/F (L/kg) |
T1/2 (hr) |
|---|---|---|---|---|---|
| 男性 | 1.9±0.3 | 130.4±27.4 | 19.9±3.9 | 1.38±0.11 | 47.1 |
| 女性 | 2.1±0.4 | 147.2±42.4 | 24.6±7.7 | 1.54±0.12 | 41.2 |
| 全体 | 2.0±0.4 | 138.8±36.1 | 22.2±6.5 | 1.46±0.14 | 44.0±12.9 |
また、HIV感染患者に本剤400mg/日を投与した場合の定常状態における血漿中濃度は4.5±1.9μg/mL(n=242)であった1),2)(外国人データ)。
16.2 吸収
16.2.1 生物学的利用率
健康成人に本剤50mgを錠剤又は内服液として単回投与し、静脈内投与時の薬物動態と比較して求めた生物学的利用率はそれぞれ93±9%、91±8%であった(外国人データ)。
16.2.2 食事及び制酸剤の影響
健康成人を対象にネビラピン200mg投与時の吸収に及ぼす食事と制酸剤の影響を検討した。その結果、食後もしくは制酸剤服用時のネビラピンの薬物動態は絶食時に比べて、吸収速度を減少させ、Tmaxの延長とCmaxのわずかな減少がみられたが、AUCには影響を及ぼさなかった(外国人データ)。
16.3 分布
健康成人男性(n=3)にネビラピン30mgを静脈内投与したときの見かけの分布容積(Vdss)は、1.21±0.09L/kgであり、広く組織に分布することが示唆された(外国人データ)。ネビラピンの血漿蛋白結合はヒト血漿中において、1~10μg/mLの濃度範囲では、約60%であった。
16.4 代謝
ヒトin vivo試験及びヒト肝ミクロゾーム試験からネビラピンはチトクロームP450により酸化的代謝を受け数種の水酸化代謝物になる。ネビラピンの酸化的代謝には、P450のCYP3A4、CYP2B6及びCYP2D6が主に関与していた。またネビラピンはそれ自身肝チトクロームP450の誘導剤であり(自己誘導)、反復投与後(200mg/日2週間投与し、その後400mg/日2週間投与)のクリアランスは単回投与時に比べ1.5から2倍に増加し、半減期は約45時間から約25~30時間に短縮した。性差はみられない(外国人データ)。[10.参照]
16.5 排泄
健康成人に本剤投与(200mg/日2週間投与し、その後400mg/日2週間投与)後、14C-ネビラピン50mgを投与し、肝代謝酵素誘導後の薬物体内動態を検討した。総放射能の91.4%が排泄され、主な代謝経路は尿(81.3%)であり、糞中には10.1%排泄された。尿中に排泄された放射能の約3%は未変化体であり、ほとんどは水酸化体とそのグルクロン酸抱合体であった(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎疾患患者
クレアチニンクリアランス値の低下が軽度(50~80mL/min)、中等度(30~50mL/min)、高度(30mL/min以下)の腎機能障害及び腎不全あるいは透析を必要とする重度腎不全の23名の腎障害患者と8名の健康成人(クレアチニンクリアランス値が80mL/min以上)において経口投与後の薬物動態を比較したとき、軽度、中等度、高度の腎機能障害患者と健康成人の間に差は見られなかった。しかし、透析腎不全患者において本剤を一週間以上投与した場合のAUCは43.5%減少し、血漿中に水酸化代謝物が増加した(外国人データ)。[9.2参照]
16.6.2 肝疾患患者
肝機能障害患者10名と健康成人8名で経口投与後の薬物動態を比較した。軽度及び中等度(Child-Pugh分類スコア7以下)の患者では本剤の投与量の調整は必要なかった。しかし、Child-Pugh分類スコア8で中等度から高度の腹水を伴う患者一人では、肝機能の悪化が本剤の血中濃度増加を招くことが示唆された(外国人データ)。[9.3参照]
16.6.3 小児
小児患者においては成人より消失が早かった(外国人データ)。[9.7参照]
16.6.4 高齢者
薬物動態に加齢の影響はみられない(外国人データ)。[9.8参照]
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第II/III相3剤併用試験
既にヌクレオシド系逆転写酵素阻害剤による治療を受けており、CD4陽性細胞数が350/mm3以下のHIV-1感染患者398例を対象にジドブジン600mg/日+ジダノシン400mg/日又はネビラピン400mg/日+ジドブジン600mg/日+ジダノシン400mg/日を二重盲検法により48週間投与した。その結果、投与40~48週におけるCD4陽性細胞数の平均変化量において3剤併用群は2剤併用群に比し有意な増加が認められた。また、HIVRNA量も同様に3剤併用群は2剤併用群に比し有意な減少を示した。
ネビラピン400mg/日+ジドブジン600mg/日+ジダノシン400mg/日群の主な副作用は発疹(8.1%)、顆粒球減少症(6.1%)、主な臨床検査異常変動項目は好中球数(14.2%)、GPT (6.1%)、GOT(4.6%)、γ-GTP(4.1%)であった3)。
17.1.2 海外第Ⅲ相3剤併用試験
未治療でCD4陽性細胞数が200~600/mm3のHIV-1感染患者151例を対象にネビラピン400mg/日+ジドブジン600mg/日+ジダノシン400mg/日、ネビラピン400mg/日+ジドブジン600mg/日もしくはジドブジン600mg/日+ジダノシン400mg/日を二重盲検法により52週間投与した。その結果、3剤併用群では投与40~52週において45%の患者でHIV RNA量が検出限界(20copies/mL)以下に減少した。この成績は他の2つの併用群に比し有意に優れていた。
ネビラピン400mg/日+ジドブジン600mg/日+ジダノシン400mg/日群の主な副作用は、嘔気(41.2%)、発疹(23.5%)、主な臨床検査異常変動項目は、γ-GTP(17.6%)、GPT(17.6%)、CPK(11.8%)であった4)。
17.2 製造販売後調査等
17.2.1 製造販売後臨床試験
HIV RNA量が1×103copies/mL以上の患者で、CD4陽性細胞数が500/mm3以下のHIV-1感染患者31例を対象に多施設共同オープン試験を実施した。投与方法は最初の2週間はネビラピン200mg/日、3週間目からは朝夕の2回にネビラピン200mgを内服し、24週間観察した。その結果、HIV RNAコピー量は投与2週間後の時点で急激に減少しそれ以降400copies/mL付近を24週まで推移した。CD4陽性細胞は投与2週間後では平均43.5%増加し、投与4週後では平均57.3%増加した。投与8週後から24週後の各時点の変化率は約81%から106%の増加であった。安全性評価対象となった31例中21例(67.7%)に副作用が認められ、主な副作用は、発熱12件(38.7%)、発疹11件(35.5%)、嘔気5件(16.1%)であった5)。
18. 薬効薬理
18.1 作用機序
ネビラピンは非ヌクレオシド系の逆転写酵素阻害剤で、HIVのタイプ1(HIV-1)の逆転写酵素を阻害し、ウイルス増殖を阻害する。ヌクレオシド系逆転写酵素阻害剤とは作用様式が異なり、核酸とは競合せず、逆転写酵素の疎水ポケット部分に結合し、逆転写酵素の触媒活性を阻害する。HIV-2逆転写酵素やヒトDNAポリメラーゼの活性は阻害しない。また本薬はヌクレオシド系逆転写酵素阻害剤に対して耐性を獲得したHIV-1の突然変異株に対しても有効であり、またヌクレオシド系逆転写酵素阻害剤やHIV-1プロテアーゼ阻害剤と併用することにより、HIV-1逆転写酵素阻害に対する相加・相乗効果が認められた6)~8)。
18.2 HIV-1増殖阻害作用
ネビラピンはHIV-1増殖に伴うヒトT細胞株c8166の細胞変性及びCD4+HeLa細胞におけるプラーク形成を阻害し、そのIC50値はそれぞれ40nM及び15nMであった6),9)(in vitro)。
18.3 免疫系及び造血系に対する影響
ネビラピンのヒトT細胞に対する細胞毒性作用9)、骨髄由来細胞の増殖抑制作用、免疫抑制作用は本薬の抗ウイルス作用に比べて極めて弱かった(in vitro)。
18.4 耐性及び交叉耐性
ネビラピンの使用によりHIV-1の耐性株が発現する。この耐性は主にHIV-1の逆転写酵素の181番目及び(または)106番目のアミノ酸の変異による10)(in vitro)。ネビラピン単独及びネビラピンとジドブジンの併用による治療を行った患者から単離したHIV-1では、治療開始直後からネビラピンに対する感受性の低下が観察され、8週間以内にすべての患者から単離したHIV-1に耐性及び変異が観察された。また、ネビラピン耐性患者から単離したHIV-1では、79%に逆転写酵素の181番目のアミノ酸に変異が認められ、それ以外にも103、106、108、188、190番目のアミノ酸に変異が認められた。一方、ネビラピンとジドブジンの両薬耐性患者から単離したHIV-1では、アミノ酸の変異パターンが異なり、181番目のアミノ酸に変異は認められず、103、106、188、190番目のアミノ酸に変異が認められた。ネビラピンとジドブジンの併用治療は、ネビラピン耐性株またはジドブジン耐性株の出現に影響を与えなかった(in vitro)。
ネビラピン+ジドブジンの2剤併用治療、ジダノシン+ジドブジンの2剤併用治療、ネビラピン+ジダノシン+ジドブジンの3剤併用治療を行った患者において、6カ月間薬物治療を行った後で血漿中からHIV-1を単離できた(HIV-1が増殖した)のは42%の患者であった。その内訳は、ネビラピン+ジドブジンが69%(11/16)、ジダノシン+ジドブジンが47%(9/19)、ネビラピン+ジダノシン+ジドブジンが21%(5/24)であり、
3剤併用した方が2剤併用よりHIV-1の増殖が観察された割合は低かった。ネビラピン+ジドブジン群及びネビラピン+ジダノシン+ジドブジン群の患者でそれぞれ増殖し単離することができたHIV-1については、すべてネビラピンに対する耐性が認められた。ネビラピンに耐性を示したHIV-1では逆転写酵素アミノ酸にK103N、Y181Cの変異が最も多く共通して認められた。なお、ジドブジンに対する耐性を持ったHIV-1が単離できた割合は、ジダノシン+ジドブジン群よりネビラピン+ジドブジン群の方が低く、ネビラピン+ジダノシン+ジドブジン群では認められなかった。また、ジダノシンに対する耐性はいずれの投与群から単離できたHIV-1においても認められなかった。すなわち、HIV-1耐性株発現の進展は、ネビラピン単剤よりも2剤併用、2剤併用よりも3剤併用治療の方が遅らせることができた。なお、ネビラピン投与によるHIV-1逆転写酵素遺伝子変異とネビラピン感受性の臨床的相関は確立されていない。
ネビラピンと他の非ヌクレオシド系逆転写酵素阻害剤との交叉耐性が観察されている。ネビラピンとヌクレオシド系逆転写酵素阻害剤やHIV-1プロテアーゼ阻害剤とは作用部位が異なり、両者の間には交叉耐性は生じにくい。ジドブジン耐性株に対してネビラピンは効果を示し、逆にネビラピン耐性株はジドブジンに対して感受性を持つことが観察されている(in vitro)。
19. 有効成分に関する理化学的知見
| 一般的名称 | ネビラピン(Nevirapine) |
|---|---|
| 化学名 | 11-cyclopropyl-5, 11-dihydro-4-methyl-6H-dipyrido[3, 2-b:2',3'-e][1, 4]diazepin-6-one |
| 分子式 | C15H14N4O |
| 分子量 | 266.30 |
| 性状 | 白色の粉末で、においはない。アセトニトリル、メタノール又はエタノール(95)に溶けにくく、水にほとんど溶けない。 |
| 化学構造式 | 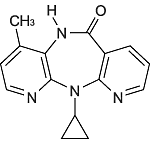 |
| 融点 | 244.7~247.5℃ |
| 分配係数 | 1.8(1-オクタノール/水) |
20. 取扱い上の注意
無包装状態の本品を高湿度(93%RH,25℃)の条件下で1カ月間保存した時、溶出率の遅延が認められているので、保存には注意すること。
22. 包装
60錠(瓶、バラ)
23. 主要文献
- Lamson M J et al:Abstract,Amer.Assoc.Pharm.Sci.Annual Meeting.Miami.FL.Nov 5-9,1995.Pharm Res.1995;12(9)(Suppl):101
- 米国添付文書
- D'Aquila R T et al:Annals of Internal Medicine.1996;124:1019-30
- Montaner J S G.et al:JAMA.1998;279(12):930-7
- 増田剛太ほか:化学療法の領域.2004;20(3):113-28
- Richman D et al:Antimicrob.Agents Chemother.1991;35(2):305-8
- Cohen K A et al:J.Biol.Chem.1991;266:14670-4
- Kopp E B et al:Nucleic Acids Res.1991;19(11):3035-9
- Merluzzi V J et al:Science.1990;250:1411-3
- Richman D et al:Proc.Natl.Acad.Sci.USA.1991;88:11241-5
24. 文献請求先及び問い合わせ先
日本ベーリンガーインゲルハイム株式会社
DIセンター
〒141-6017 東京都品川区大崎2丁目1番1号
ThinkPark Tower
0120-189-779
(受付時間)9:00~18:00
(土・日・祝日・弊社休業日を除く)
26. 製造販売業者等
26.1 製造販売元(輸入販売元)
日本ベーリンガーインゲルハイム株式会社
東京都品川区大崎2丁目1番1号