カレトラ配合内用液の添付文書
- 商品名:
- カレトラ配合内用液
- 一般名:
- ロピナビル+リトナビル
- 略称 :
- LPV/rtv

添付文書の読み方
ここで提供している添付文書情報は、2025年3月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
抗ウイルス化学療法剤
ロピナビル+リトナビル
- ** 2024年6月改訂(第5版)
* 2023年8月改訂(第4版) - 規制区分:劇薬、処方箋医薬品注)
注)注意-医師等の処方箋により使用すること
| 日本標準商品分類番号 | 87625 |
|---|
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 2年 |
| 承認番号 | 22100AMX00434000 |
|---|---|
| 販売開始 | 2000年12月 |
2. 禁忌(次の患者には投与しないこと)
2.1
本剤の成分に対し過敏症の既往歴のある患者
2.2
次の薬剤を投与中の患者:ピモジド、エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン、ジヒドロエルゴタミンメシル酸塩、エルゴメトリンマレイン酸塩、メチルエルゴメトリンマレイン酸塩、ミダゾラム、トリアゾラム、ルラシドン塩酸塩、バルデナフィル塩酸塩水和物、シルデナフィルクエン酸塩(レバチオ)、タダラフィル(アドシルカ)、ブロナンセリン、アゼルニジピン、アゼルニジピン・オルメサルタン メドキソミル、リバーロキサバン、ロミタピドメシル酸塩、ベネトクラクス〈再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)の用量漸増期〉、ボリコナゾール、グラゾプレビル水和物[10.1 参照]
2.3
腎機能又は肝機能障害のある患者で、コルヒチンを投与中の患者[9.2.1 参照],[9.3.1 参照],[10.2 参照]
3. 組成・性状
3.1 組成
| 有効成分 | 1mL中 ロピナビル80mg・リトナビル20mg |
| 添加剤 | エタノール、プロピレングリコール、サッカリンナトリウム水和物、ポリオキシエチレン硬化ヒマシ油40、ポビドン、グリセリン、トウモロコシシロップ、塩化ナトリウム、クエン酸ナトリウム水和物、アセスルファムカリウム、無水クエン酸、l-メントール、ベンジルアルコール、バニリン、香料 |
3.2 製剤の性状
| 色・剤形 | 淡黄色~だいだい色の澄明な液体 |
|---|
4. 効能又は効果
HIV-1感染症
6. 用法及び用量
通常、成人にはロピナビル・リトナビルとして1回400mg・100mg(5mL)を1日2回食後に経口投与する。
通常、小児には、体重7kg以上15kg未満で1kgあたり12mg・3mg、15kg以上40kg以下で1kgあたり10mg・2.5mgを1日2回食後に経口投与する。最大投与量は400mg・100mg(5mL)1日2回投与とする。
7. 用法及び用量に関連する注意
本剤の吸収を高めるため、食後に服用すること。[16.2.1 参照]
8. 重要な基本的注意
* 8.1
本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又はそれに代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。
8.1.1
本剤はHIV感染症の根本的治療薬ではないことから、日和見感染を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。
8.1.2
本剤の長期投与による影響については、現在のところ不明であること。
8.1.3
本剤投与開始後、担当医の指示なしに用量を変更したり、服用を中止したりしないこと。
8.1.4
本剤は併用薬剤と相互作用を起こすことがあるため、服用中のすべての薬剤を担当医に報告すること。また、本剤で治療中に新たに他の薬剤を服用する場合、事前に担当医に相談すること。[10.、16.7.1、16.7.2参照]
8.1.5
本剤はエタノール42.4%を含有する。本剤の成人1日用量(10mL)ではエタノール約4.3mLに相当するので、自動車の運転等危険を伴う作業をする際には注意すること。[9.7.2 参照],[10.2 参照],[13.2 参照]
8.2
HIVプロテアーゼ阻害薬にて治療中の患者において糖尿病の発症や悪化、もしくは高脂血症(コレステロール、トリグリセリドの上昇)が報告されているので、定期的な検査等を行うこと。[11.1.1 参照]
8.3
本剤の使用例で著しいトリグリセリド上昇を伴う膵炎が報告されている。血清リパーゼ、アミラーゼ、トリグリセリド等の定期的な検査を行うこと。[11.1.2 参照]
8.4
使用期間の長短を問わず定期的な肝機能検査値等の測定を行い、観察を十分に行うこと。[9.3.2、16.6.1参照]
8.5
本剤を含む抗HIV薬の多剤併用療法を行った患者で、免疫再構築症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 血友病及び著しい出血傾向を有する患者
HIVプロテアーゼ阻害薬にて治療中の血友病の患者において突発性の出血性関節症をはじめとする出血事象の増加が報告されている。[11.1.3 参照]
9.1.2 器質的心疾患及び心伝導障害(房室ブロック等)のある患者
本剤は軽度の無症候性PR間隔の延長が認められている。[10.2、17.3.1参照]
9.1.3 B型肝炎、C型肝炎を合併している患者
肝機能障害を増悪させるおそれがある。
9.2 腎機能障害患者
9.2.1 腎機能障害のある患者で、コルヒチンを投与中の患者
投与しないこと。コルヒチンの血中濃度が上昇するおそれがある。[2.3、10.2 参照]
9.3 肝機能障害患者
9.3.1 肝機能障害のある患者で、コルヒチンを投与中の患者
投与しないこと。コルヒチンの血中濃度が上昇するおそれがある。[2.3、10.2 参照]
9.3.2 肝機能障害のある患者(コルヒチンを投与中の患者を除く)
定期的に肝機能検査値や薬物血中濃度測定等を行い、慎重に投与すること。本剤は主に肝臓で代謝されるため、高い血中濃度が持続するおそれがある。また、トランスアミナーゼの上昇を合併している患者では肝機能障害を増悪させるおそれがある。[8.4、16.6.1 参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験(ラット)では、ロピナビルとリトナビル(2:1)を最大耐量で投与し、推奨臨床用量で到達しうる濃度よりやや低い血中濃度に到達させたが、妊孕性への影響は認めなかった。
妊娠動物(ラット及びウサギ)にロピナビル・リトナビルを投与した試験では、投与に関連した形成異常を認めなかった。ラットにおける検討では、母動物に毒性があらわれる用量(100mg/kg/日・50mg/kg/日)において、発生毒性(吸収胚、胎児生存率の低下、胎児体重の低下、骨格変異及び骨化遅延の発現率上昇)が認められた。周産期ラットにおいては発生毒性(生後21日目までの胎児生存率低下)が認められた。ウサギにおける検討では、母動物に毒性があらわれる用量(80mg/kg/日・40mg/kg/日)において、発生毒性を認めなかった。
9.6 授乳婦
授乳を避けさせること。米国疾病管理センター(CDC)は、HIV伝播を避けるため、HIV陽性の母親は授乳を避けるよう勧告している。ロピナビルは乳汁に移行することが報告されている(ラット)。ヒト乳汁への本剤の移行は不明である。
9.7 小児等
9.7.1
12歳未満のHIV感染症小児を対象とした臨床試験は実施していないが、有害事象の発生状況においては成人との差は認められていない。
9.7.2
特に新生児や乳児においては本剤に含有されるエタノールやプロピレングリコール(エタノールはプロピレングリコールの代謝を阻害する)の代謝能が低い。[8.1.5 参照],[10.2 参照],[13.2 参照]
9.8 高齢者
生理機能の低下及び合併症、併用薬剤等に注意すること。高齢者における薬物動態については十分な検討がなされていない。
10. 相互作用
本剤は肝チトクロームP450(CYP)のアイソザイムであるCYP3Aとの親和性が強い(in vitro)。主にCYP3Aで代謝される薬剤を本剤と併用することにより、併用薬剤の代謝を競合的に阻害し、併用薬剤の血中濃度を上昇させることがある。一方でCYP3Aを誘導する薬剤を本剤と併用すると、本剤の血中濃度が低下することがある。また、CYP3Aを阻害する薬剤との併用で本剤の血中濃度が上昇することがある。
他の薬剤との相互作用は、可能なすべての組み合わせについて検討されているわけではないので、併用に際しては用量に留意して慎重に投与すること。[8.1.4、16.4、16.7.1、16.7.2参照]
** 10.1 併用禁忌(併用しないこと)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
|
ピモジド [2.2参照] |
不整脈のような重篤な又は生命に危険を及ぼすような事象を起こすおそれがある。 | 本剤のチトクロームP450に対する競合的阻害作用により、併用した場合これらの薬剤の血中濃度が大幅に上昇することが予測される。 |
エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン
エルゴメトリンマレイン酸塩
|
血管攣縮などの重篤な又は生命に危険を及ぼすような事象を起こすおそれがある。 | |
ミダゾラム
|
過度の鎮静や呼吸抑制を起こすおそれがある。 | |
バルデナフィル塩酸塩水和物
|
低血圧などの重篤な又は生命に危険を及ぼすような事象を起こすおそれがある。 | |
ブロナンセリン
|
これら薬剤の血中濃度上昇により、重篤な又は生命に危険を及ぼすような事象を起こすおそれがある。 | |
ベネトクラクス
〈再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)の用量漸増期〉
|
ベネトクラクスの再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)の用量漸増期に本剤を併用した場合、腫瘍崩壊症候群の発現が増強されるおそれがある。 | 本剤がCYP3Aによるベネトクラクスの代謝を競合的に阻害するため。 |
ボリコナゾール
|
リトナビルとの併用でボリコナゾールの血中濃度が低下したとの報告がある。 | リトナビルのチトクロームP450の誘導作用によるものと考えられている。 |
グラゾプレビル水和物
|
グラゾプレビルの血中濃度が上昇したとの報告がある。 | ロピナビルのOATP1B阻害作用によるものと考えられている。 |
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
シルデナフィルクエン酸塩
|
これら薬剤の血中濃度が上昇し、低血圧、失神、視覚障害や勃起持続等のこれら薬剤の副作用が発現するおそれがある。 | 本剤がCYP3Aによるこれら薬剤の代謝を競合的に阻害するため。 |
|
シンバスタチン アトルバスタチンカルシウム水和物 [16.7.2参照] |
これら薬剤の血中濃度が上昇し、これら薬剤の副作用が発現しやすくなるおそれがある。特にシンバスタチンとの併用はなるべく避けること。 | |
|
イトラコナゾール ケトコナゾール※ [16.7.2参照] |
これら薬剤の血中濃度が上昇するおそれがある。高用量(200mg/日をこえる)投与は避けること。 | |
|
ジヒドロピリジン骨格を有するCa拮抗剤 (フェロジピン、ニフェジピン、ニカルジピン塩酸塩等) リファブチン サルメテロールキシナホ酸塩 ダサチニブ ニロチニブ ビンカアルカロイド系抗悪性腫瘍剤 (ビンブラスチン硫酸塩、ビンクリスチン硫酸塩等) ボセンタン水和物 コルヒチン クエチアピンフマル酸塩 シメプレビルナトリウム [2.3、9.2.1、9.3.1、16.7.2参照] |
これら薬剤の血中濃度が上昇し、これら薬剤の副作用が発現しやすくなるおそれがある。 | |
| クラリスロマイシン | 腎機能障害のある患者ではクラリスロマイシンの血中濃度が上昇するおそれがある。 | |
|
シクロスポリン タクロリムス水和物 エベロリムス **シロリムス1) |
これら薬剤の血中濃度が上昇するおそれがある。治療域のモニタリングを行うことが望ましい。 | |
| トラゾドン塩酸塩 | トラゾドンの血中濃度が上昇し、副作用が発現しやすくなるおそれがある。トラゾドンの減量を考慮すること。 | |
|
フルチカゾンプロピオン酸エステル ブデソニド トリアムシノロンアセトニド |
これら薬剤の血中濃度が上昇するおそれがある。これら薬剤との併用において、クッシング症候群、副腎皮質機能抑制等が報告されているので、併用は治療上の有益性がこれらの症状発現の危険性を上回ると判断される場合に限ること。 | |
|
フェンタニル フェンタニルクエン酸塩 |
フェンタニルの血中濃度が上昇し、副作用が発現しやすくなるおそれがある。副作用(呼吸抑制等)に対する十分なモニタリングを行うことが望ましい。 | |
|
イブルチニブ エンコラフェニブ |
これら薬剤の血中濃度が上昇し、副作用が増強されるおそれがある。本剤からCYP3A阻害作用のない薬剤への代替を考慮すること。やむを得ず併用する際には、これら薬剤の減量を考慮するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | |
|
ベネトクラクス 〈再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)の維持投与期、急性骨髄性白血病〉 |
ベネトクラクスの再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)の維持投与期又は急性骨髄性白血病に対してベネトクラクス投与中に本剤を併用した場合、ベネトクラクスの副作用が増強されるおそれがあるので、ベネトクラクスを減量するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | |
| アパルタミド | アパルタミドの血中濃度が上昇し、副作用が増強されるおそれがある。 また、本剤の血中濃度が減少するおそれがある。本剤からCYP3A阻害作用のない薬剤への代替を考慮すること。やむを得ず併用する際には、アパルタミドの減量を考慮するとともに、患者の状態を慎重に観察し、副作用の発現や本剤の効果の減弱に十分注意すること。 |
本剤がCYP3Aによるアパルタミドの代謝を競合的に阻害するため。 また、アパルタミドがCYP3Aを誘導するため。 |
| リオシグアト | リオシグアトの血中濃度が上昇するおそれがある。本剤との併用が必要な場合は、患者の状態に注意し、必要に応じてリオシグアトの減量を考慮すること。 | 本剤のCYP1A1及びCYP3A阻害によりリオシグアトのクリアランスが低下する。 |
|
アミオダロン塩酸塩 ベプリジル塩酸塩水和物 リドカイン塩酸塩 キニジン硫酸塩水和物 フレカイニド酢酸塩 プロパフェノン塩酸塩 |
これら薬剤の血中濃度が上昇するおそれがある。血中濃度のモニタリングを行うことが望ましい。 | 本剤が肝薬物代謝酵素によるこれら薬剤の代謝を競合的に阻害するためと考えられている。 |
| ジゴキシン | ジゴキシンの血中濃度が上昇するおそれがある。血中濃度のモニタリングを行うことが望ましい。 | リトナビルのP-gp阻害作用によるものと考えられている。 |
| アファチニブマレイン酸塩 | アファチニブの血中濃度が上昇し、副作用が発現しやすくなるおそれがある。本剤はアファチニブと同時かアファチニブ投与後に投与すること。 | |
| ロスバスタチンカルシウム | ロスバスタチンの血中濃度が上昇し、ロスバスタチンの副作用が発現しやすくなるおそれがある。 | 主としてロピナビルのOATP1B1阻害作用によるものと考えられている。リトナビルのBCRP阻害作用も関与している可能性がある。 |
| グレカプレビル・ピブレンタスビル | グレカプレビル及びピブレンタスビルの血中濃度が上昇するおそれがある。 | 本剤のOATP1B、P-gp又はBCRP阻害作用によるものと考えられる。 |
| セイヨウオトギリソウ(St. John's Wort、セント・ジョーンズ・ワート)含有食品 | 本剤の代謝が促進され血中濃度が低下するおそれがあるので、本剤投与時はセイヨウオトギリソウ含有食品を摂取しないよう注意すること。 | セイヨウオトギリソウにより誘導された肝薬物代謝酵素(チトクロームP450)が本剤の代謝を促進し、クリアランスを上昇させるためと考えられている。 |
|
リファンピシン [16.7.2参照] |
本剤の血中濃度が低下し、治療効果を減弱させるおそれがある。併用はなるべく避けること。 | これら薬剤がCYP3Aを誘導するため。 |
|
カルバマゼピン フェノバルビタール デキサメタゾン |
ロピナビルの血中濃度が低下するおそれがある。 | |
| フェニトイン | ロピナビル及びフェニトインの血中濃度が低下するおそれがある。 | 相互に肝薬物代謝酵素を誘導するためと考えられている。 |
| ワルファリンカリウム | ワルファリンの血中濃度に影響を与えることがある。INRのモニタリングを行うことが望ましい。 | 肝薬物代謝酵素の関与が考えられるが機序不明。 |
| エルバスビル | エルバスビルの血中濃度が上昇したとの報告がある。 | |
|
エチニルエストラジオール エストラジオール安息香酸エステル [16.7.2参照] |
これら薬剤の血中濃度が低下するおそれがある。 エストロゲンをベースとする避妊剤と併用する場合は、他の避妊法に変更するか避妊法を追加する必要がある。 |
本剤がこれら薬剤の肝薬物代謝酵素を誘導するためと考えられている。 |
|
ラモトリギン バルプロ酸ナトリウム [16.7.2参照] |
これら薬剤の血中濃度が低下するおそれがある。 | 本剤がグルクロン酸抱合を促進するためと考えられている。 |
|
メサドン塩酸塩 [16.7.2参照] |
機序不明 | |
|
ジスルフィラム様作用を有する薬剤 ジスルフィラム、シアナミド、メトロニダゾール 等 [8.1.5 参照],[9.7.2 参照] |
ジスルフィラムあるいはシアナミド-アルコール反応を起こすおそれがある。 | 本剤はエタノール42.4%を含有するため。 |
|
PR間隔を延長させる薬剤 ベラパミル塩酸塩 アタザナビル硫酸塩 等 [9.1.2、17.3.1参照] |
PR間隔が延長するおそれがある。 | 本剤は軽度の無症候性PR間隔の延長が認められている。 |
|
ジドブジン アバカビル硫酸塩 |
これら薬剤の血中濃度を低下させるおそれがある。臨床的な影響は不明である。 | 本剤がグルクロン酸抱合を誘導するためと考えられている。 |
| テノホビル | テノホビルの血中濃度が上昇し、腎機能障害等の副作用があらわれやすくなるおそれがある。 | 機序不明 |
|
マラビロク [16.7.2参照] |
マラビロクの血中濃度が上昇するおそれがある。 | 本剤がCYP3Aにおけるこれら薬剤の代謝を競合的に阻害するため。 |
| リルピビリン塩酸塩 | リルピビリンの血中濃度が上昇したとの報告がある。リルピビリンの用量調節の必要性は認められていない。 | |
|
ネルフィナビル [7. 参照],[16.7.2 参照],[16.7.3 参照] |
ネルフィナビルの血中濃度が上昇するおそれがある。 ロピナビルの血中濃度が低下するおそれがある。 |
本剤がCYP3Aにおけるネルフィナビルの代謝を競合的に阻害するため。 ロピナビル血中濃度低下の機序は不明。 |
|
ネビラピン エファビレンツ [7. 参照],[16.7.2 参照],[16.7.3 参照] |
ロピナビルの血中濃度が低下するおそれがある。 | これら薬剤がCYP3Aを誘導するため。 |
| エトラビリン | エトラビリンの血中濃度が低下したとの報告がある。エトラビリンの用量調節の必要性は認められていない。 | リトナビルの肝薬物代謝酵素誘導作用によるものと考えられている。 |
|
ホスアンプレナビル [7.2、16.7.3参照] |
アンプレナビルの血中濃度が低下するおそれがある。 併用に関する推奨用量は確立されていない。 |
肝薬物代謝酵素の関与が考えられるが機序不明。 |
| ** ホスタマチニブナトリウム水和物 |
ホスタマチニブの活性代謝物であるR406の血中濃度が上昇し、副作用が増強されるおそれがある。 併用時には患者の状態を慎重に観察して副作用の発現に十分注意し、必要に応じてホスタマチニブの減量を考慮すること。 |
本剤がCYP3Aにおけるホスタマチニブの代謝を競合的に阻害するためと考えられている。 |
※経口剤は国内未発売
11. 副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1 重大な副作用
11.1.1 高血糖、糖尿病(いずれも頻度不明)
高血糖、糖尿病及び糖尿病の悪化があらわれることがある。HIVプロテアーゼ阻害薬にて治療中の患者に糖尿病、糖尿病の悪化及び高血糖があらわれたとの報告がある。一部の例ではインスリン又は経口糖尿病薬の投与開始や用量調節が必要となった。一部では糖尿病性ケトアシドーシスがあらわれている。HIVプロテアーゼ阻害薬を中止した例の一部では、高血糖が持続した。[8.2参照]
11.1.2 膵炎(頻度不明)
嘔気、嘔吐、腹痛等の臨床症状や血清リパーゼ、アミラーゼ、トリグリセリド等の検査値異常があらわれた場合は膵炎を疑うこと。[8.3参照]
11.1.3 出血傾向(頻度不明)
出血事象があらわれた場合には血液凝固因子を投与するなど適切な処置を行うこと。[9.1.1参照]
11.1.4 肝機能障害、肝炎(いずれも頻度不明)
11.1.5 徐脈性不整脈(頻度不明)
洞徐脈、洞停止、房室ブロックがあらわれることがある。
11.1.6 中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)、皮膚粘膜眼症候群(Stevens-Johnson症候群)、多形紅斑(いずれも頻度不明)
11.2 その他の副作用
| 2%以上 | 2%未満 | 頻度不明 | |
|---|---|---|---|
| 全身症状 | 頭痛 | 無力症、疼痛、背部痛、胸痛、悪寒、嚢胞、浮腫、末梢性浮腫、顔面浮腫、発熱、インフルエンザ、倦怠感、ウイルス感染、細菌感染、過敏症、肥大、薬物過敏症、免疫再構築症候群、四肢痛、顔面腫脹 | 体脂肪の再分布/蓄積(胸部、体幹部の脂肪増加、末梢部の脂肪減少、野牛肩) |
| 循環器 | − | 深部静脈血栓症、高血圧、心悸亢進、血栓性静脈炎、血管炎、血管障害、心房細動、起立性低血圧、静脈瘤、心筋梗塞、血管拡張、狭心症、三尖弁閉鎖不全症 | − |
| 消化器 | 下痢、嘔気、腹痛、嘔吐、アミラーゼ上昇、鼓腸 | 消化不良、食欲不振、胆嚢炎、便秘、口内乾燥、嚥下障害、腸炎、おくび、食道炎、大便失禁、胃炎、胃腸炎、出血性腸炎、食欲亢進、唾液腺炎、口内炎、潰瘍性口内炎、異常便、腹部膨満感、小腸炎、歯周炎、胆管炎、上腹部痛、リパーゼ上昇、腹部不快感、下腹部痛、十二指腸炎、胃潰瘍、胃食道逆流性疾患、痔核、直腸出血 | − |
| 肝臓 | 肝機能検査異常、ビリルビン値上昇 | 黄疸、肝腫大 | − |
| 血液 | 血小板減少、好中球減少 | 貧血、白血球減少症、リンパ節症、脾腫、ヘモグロビン減少 | − |
| 代謝・栄養 | 総コレステロール上昇、トリグリセリド上昇、ナトリウム低下、ナトリウム上昇 | ビタミン欠乏症、脱水、耐糖能低下、乳酸性アシドーシス、肥満、体重減少、血中尿酸上昇、無機リン低下、CK上昇 | − |
| 内分泌系 | − | クッシング症候群、甲状腺機能低下、女性型乳房、乳房腫大 | − |
| 筋骨格 | − | 筋肉痛、関節痛、骨関節炎、骨壊死 | − |
| 精神神経系 | − | 不眠、異夢、激越、健忘、不安、運動失調、錯乱状態、抑うつ、浮動性めまい、回転性めまい、ジスキネジア、感情不安定、脳症、緊張亢進、リビドー減退、神経過敏、ニューロパチー、末梢性ニューロパチー、感覚異常、末梢神経炎、傾眠、思考異常、振戦、無感情、脳梗塞、痙攣、顔面神経麻痺、片頭痛、錐体外路症状、失見当識、気分動揺、平衡障害 | − |
| 皮膚 | − | 発疹、ざ瘡、脱毛、皮膚乾燥、剥脱性皮膚炎、せつ腫症、斑状丘疹性皮疹、爪疾患、そう痒、良性皮膚腫瘍、皮膚変色、多汗症、湿疹、脂漏、皮膚潰瘍、蜂巣炎、毛包炎、脂肪腫症、アレルギー性皮膚炎、特発性毛細血管炎、皮膚肥厚 | − |
| 呼吸器 | − | 呼吸困難、肺水腫、副鼻腔炎、咽頭炎、喘息、鼻炎、気管支炎、気管支肺炎 | − |
| 感覚器 | − | 視覚障害、眼疾患、中耳炎、味覚異常、耳鳴、聴覚過敏 | − |
| 泌尿器・生殖器 | − | 射精障害、男性性腺機能低下、腎結石、尿異常、腎炎、無月経、会陰膿瘍、血尿、尿臭異常、月経過多、クレアチニンクリアランス低下 | − |
13. 過量投与
13.1 処置
本剤の過量投与では、急性アルコール中毒を起こす可能性がある。
13.2 処置
有効成分ロピナビル・リトナビルの蛋白結合率が高いため、透析による除去効果は低い。本剤に含有されるエタノール及びプロピレングリコールは透析によって除去できる。[8.1.5 参照],[9.7.2 参照]
14. 適用上の注意
14.1 薬剤交付時の注意
本剤は、冷蔵庫内(2~8℃)で保存すること。なお、携帯の目的で一時的に冷蔵庫外に出す場合、25℃以上を避けること。
15. その他の注意
15.1 臨床使用に基づく情報
HIVプロテアーゼ阻害薬(特に逆転写酵素阻害薬との併用例において)によりCK上昇、筋肉痛、筋炎、まれに横紋筋融解症を発現したとの報告がある。
15.2 非臨床試験に基づく情報
長期がん原性試験で、最大耐量(通常用量 ロピナビル・リトナビル400/100mg1日2回投与のヒトにおけるロピナビルの曝露量のほぼ2倍)を投与したマウスで肝腫瘍の軽度の増加が認められている。
16. 薬物動態
16.1 血中濃度
16.1.1 反復投与
〈HIV陽性患者〉
ロピナビル・リトナビル400mg・100mg BIDを投与したHIV陽性患者におけるロピナビルの定常期血中濃度は、リトナビル濃度の15~20倍であった。(ロピナビルの各パラメータ:AUC=160μg・hr/mL、Cmax=9.58±4.41μg/mL、Cmin=3.83±3.44μg/mL、tmax=3±2hr、T1/2=平均5~6時間、見かけの経口クリアランス(CL/F)=6.4±4.4L/hr)リトナビルの血中濃度は、リトナビル600mg BIDを投与した場合の血中濃度の7%未満であった。ロピナビルのin vitro EC50は、リトナビルの約10分の1である。
HIV陽性成人患者21名に対し、ロピナビル・リトナビル400mg・100mgを1日2回、3~4週間にわたり投与した場合のロピナビルとリトナビルの定常状態血中濃度の平均値を下図に示す(外国人データ)。
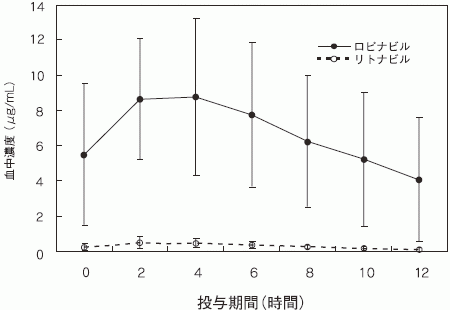
〈健康被験者及びHIV感染症患者〉
男性健康被験者及びHIV感染症患者に対しリトナビルの併用の有無によりロピナビルの薬物動態を検討したところ、健康被験者−患者間で差を認めなかった。健康被験者及びHIV感染症患者を対象とした複数の臨床試験ではロピナビルの血中濃度は投与開始から約10~14日で定常状態に到達した(外国人データ)。
16.2 吸収
16.2.1 食事が経口投与に及ぼす影響
健康被験者で脂肪含量が中等度の食事(500~682kcal、22.7~25.1%は脂肪由来)の後にロピナビル・リトナビル3カプセル(400mg・100mg)を単回投与した場合、ロピナビルのAUCが48%、Cmaxが23%上昇した(空腹時投与との比較)。高脂肪食(872kcal、55.8%が脂肪由来)の摂取後のカプセル剤投与ではAUCは97%、Cmaxは43%上昇した(空腹時投与との比較)。本剤は食後投与すること(外国人データ)。[7. 参照]
16.3 分布
定常期におけるロピナビルの血漿蛋白結合率は約98~99%(血漿遊離分画(Fu):約1~2%)である。ロピナビルは、α1-酸性糖蛋白質(AAG)とアルブミンに結合するが、親和性はAAGの方が高い2)。ロピナビル・リトナビル400mg・100mg BIDの投与後に認められる濃度範囲では、定常期におけるロピナビルの血漿蛋白結合率は一定であり、健康被験者とHIV陽性患者との間に差は認められていない(外国人データ)。
16.4 代謝
ヒト肝ミクロソームを用いたin vitro試験で、ロピナビルは主に酸化代謝を受けることが示された。ロピナビルはCYPのアイソザイムのうち、主としてCYP3Aにより代謝される。リトナビルはCYP3Aと強い親和性を示し、CYP3Aによるロピナビルの代謝を阻害するためロピナビルの血中濃度が上昇する。健康被験者に14C標識ロピナビルを用いたロピナビル・リトナビル400mg・100mgを単回投与した場合、血中放射活性の89%が未変化体に由来した。ロピナビルの酸化代謝物は、ヒトでは少なくとも13種類認められている。4-oxo体及び4-水酸化体のエピマー各2種が抗ウイルス活性をもつ代謝物であるが、その量は血中の総放射活性物量のごく一部である。リトナビルは代謝酵素を誘導して自らの代謝を誘導するため、ロピナビルの代謝も誘導すると考えられる3) 4)。[10.、16.7.1参照]
16.5 排泄
健康被験者に14C標識ロピナビル・リトナビルの400mg・100mgを単回経口投与した場合、10.4±2.3%が尿中へ、82.6±2.5%が糞中へ排泄された。また、未変化体約2.2%が尿中へ、19.8%が糞中へ排泄された(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 軽度~中等度の肝機能障害患者
HIVとHCVに感染している軽度~中等度の肝機能障害患者(n=12)と肝機能障害のないHIV感染症患者(n=12)に対する薬物動態臨床試験(ロピナビル・リトナビル400mg・100mg BID)において、肝機能障害患者群では非肝機能障害患者群と比較して、ロピナビルのAUCが約30%、Cmaxが約20%上昇し、蛋白結合率は低下した(HIV・HCV感染患者:99.09%、HIV・非肝機能障害患者:99.31%)。なお、重度の肝機能障害患者における臨床試験は行われていない5)。[8.4、9.3.2参照]
16.7 薬物相互作用
16.7.1 in vitro試験
本剤は、主としてCYP3Aにより代謝される。本剤に含まれるリトナビルはCYP3Aと特に強い親和性を示し、主にCYP3A(3A4、3A5、3A7)で代謝される薬剤の代謝を競合的に阻害する。臨床用量で得られる濃度の範囲ではCYP2D6、CYP2C9、CYP2C19、CYP2E1、CYP2B6、CYP1A2を阻害しない。[8.1.6、10.、16.4参照]
16.7.2 薬物相互作用臨床試験
本剤と併用する可能性の高い薬剤について、それら薬剤の薬物動態への影響を以下に示す。[8.1.6、10.、10.2参照]
| 併用薬 | 併用薬の |
ロピナビル・ |
n | ロピナビル薬物動態の |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| アンプレナビル | 750BID、10日 | 400・100 BID、21日 | 12 | 0.72 | 0.62 | 0.43 |
| アトルバスタチン | 20QD、4日 | 400・100 BID、14日 | 12 | 0.90 | 0.90 | 0.92 |
| エファビレンツ1 | 600QHS、9日 | 400・100 BID、9日 | 11, 7* | 0.97 | 0.81 | 0.61 |
| ケトコナゾール | 200単回 | 400・100 BID、16日 | 12 | 0.89 | 0.87 | 0.75 |
| ネルフィナビル | 1000BID、10日 | 400・100 BID、21日 | 13 | 0.79 | 0.73 | 0.62 |
| ネビラピン | 200BID、定常(1年以上)2 | 400・100 BID、定常(1年以上) | 22, 19* | 0.81 | 0.73 | 0.49 |
| 7mg/kgもしくは4mg/kgQD、2週;BID1週3 | 300・75mg/m2 BID、3週 | 12, 15* | 0.86 | 0.78 | 0.45 | |
| オメプラゾール | 40QD、5日 | 400・100 BID†、10日 | 12 | 1.08 | 1.07 | 1.03 |
| 800・200 QD†、10日 | 12 | 0.94 | 0.92 | 0.71 | ||
| ラニチジン | 150単回 | 400・100 BID†、10日 | 12 | 0.98 | 0.98 | 0.93 |
| 800・200 QD†、10日 | 11 | 0.98 | 0.96 | 0.85 | ||
| プラバスタチン | 20QD、4日 | 400・100 BID、14日 | 12 | 0.98 | 0.95 | 0.88 |
| リファブチン | 150QD、10日 | 400・100 BID、20日 | 14 | 1.08 | 1.17 | 1.20 |
| リファンピシン6 | 600QD、10日 | 400・100 BID、20日 | 22 | 0.45 | 0.25 | 0.01 |
| 600QD、14日 | 800・200 BID、9日4 | 10 | 1.02 | 0.84 | 0.43 | |
| 600QD、14日 | 400・400 BID、9日5 | 9 | 0.93 | 0.98 | 1.03 | |
| リトナビル2 | 100BID、 3-4週 |
400・100 BID、3-4週 | 8, 21* | 1.28 | 1.46 | 2.16 |
| テラプレビル | 750TID、10日 | 400・100 BID、20日 | 12 | 0.96 | 1.06 | 1.14 |
特に断りのない限りすべて健康被験者におけるカプセル剤又は液剤の試験である
- リトナビルの薬物動態はエファビレンツ併用の影響を受けない
- HIV陽性成人患者の試験
- HIV陽性患児(6ヵ月齢~12歳)の試験
- 漸増投与800・200BID(533・133BID×1日、667・167BID×1日、800・200BID×7日)と400・100 BID×10日との比較
- 漸増投与400・400BID(400・200BID×1日、400・300BID×1日、400・400BID×7日)と400・100 BID×10日との比較
- 標準用量の本剤との併用は推奨されない
* 平行法による検討(n:ロピナビル・リトナビル+併用薬投与例、ロピナビル・リトナビル単独投与例)
† 錠剤による試験
注)本剤の承認最大用量は400mg・100mgを1日2回投与である。
| 併用薬 | 併用薬の |
ロピナビル・ |
n | 併用薬の |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| アンプレナビル1 | 750BID、10日併用 対 1200BID、14日単独 |
400・100 BID、21日 | 11 | 1.12 | 1.72 | 4.57 |
| アトルバスタチン | 20QD、4日 | 400・100 BID、14日 | 12 | 4.67 | 5.88 | 2.28 |
| エファビレンツ | 600QHS、9日 | 400・100 BID、9日 | 11, 12* | 0.91 | 0.84 | 0.84 |
| エチニルエストラジオール | 35μgQD、21日 | 400・100 BID、14日 | 12 | 0.59 | 0.58 | 0.42 |
| インジナビル1 | 600BID、10日併用/食後 対 800TID、5日単独/空腹 |
400・100 BID、15日 | 13 | 0.71 | 0.91 | 3.47 |
| ケトコナゾール | 200単回 | 400・100 BID、16日 | 12 | 1.13 | 3.04 | N/A |
| ラモトリギン | 100BID、12日対 100BID、8日単独 |
400・100 BID、12日 | 18 | 0.54 | 0.5 | 0.44 |
| 200BID、9日対 100BID、8日単独 |
400・100 BID、9日 | 15 | 1.03 | 0.91 | 0.79 | |
| マラビロク | 300BID | 400・100 BID | 11 | 1.97 | 3.95 | 9.24 |
| メサドン | 5単回 | 400・100 BID、10日 | 11 | 0.55 | 0.47 | N/A |
| ネルフィナビル1 | 1000BID、10日併用 対 1250BID、14日単独 |
400・100 BID、21日 | 13 | 0.93 | 1.07 | 1.86 |
| M8代謝物 | 2.36 | 3.46 | 7.49 | |||
| ネビラピン | 200QD、14日;200BID、6日 | 400・100 BID、20日 | 5, 6* | 1.05 | 1.08 | 1.15 |
| ノルエチンドロン | 1QD、21日 | 400・100 BID、14日 | 12 | 0.84 | 0.83 | 0.68 |
| プラバスタチン | 20QD、4日 | 400・100 BID、14日 | 12 | 1.26 | 1.33 | N/A |
| リファブチン | 150QD、10日併用 対 300QD、10日単独 |
400・100 BID、10日 | 12 | 2.12 | 3.03 | 4.90 |
| 25-O脱アセチルリファブチン | 23.6 | 47.5 | 94.9 | |||
| リファブチン+25-O脱アセチルリファブチン2 | 3.46 | 5.73 | 9.53 | |||
| テラプレビル | 750TID、10日 | 400・100 BID、20日 | 12 | 0.47 | 0.46 | 0.48 |
| サキナビル1 | 800BID、10日併用 対 1200TID、5日単独 |
400・100 BID、15日 | 14 | 6.34 | 9.62 | 16.74 |
| 1200BID、5日併用 対 1200TID、5日単独 |
400・100 BID、20日 | 10 | 6.44 | 9.91 | 16.54 | |
特に断りのない限りすべて健康被験者におけるカプセル剤又は液剤の試験である
- 1.用量補正は行っていない
- 2.用量補正後の合計
* 平行法による検討(n:ロピナビル・リトナビル+併用薬投与例、併用薬単独投与例)
N/A:データなし
16.7.3 他の抗HIV薬との併用
ラルテグラビル:臨床的に影響のある相互作用は認められていない(外国人データ)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第III相試験
試験863(カプセル剤):抗HIV薬治療未経験の成人HIV感染症患者653例を対象として、ロピナビル・リトナビル(LPV/r)400mg・100mg BID、サニルブジン(d4T)及びラミブジン(3TC)の併用群、もしくはネルフィナビル(NFV)750mgTID、サニルブジン及びラミブジンの併用群に無作為に割り付け、多施設二重盲検試験を実施した。開始時の平均CD4リンパ球数は259cells/mm3(2~949cells/mm3)で、平均血中HIV RNA量は4.9log10copies/mL(2.6~6.8log10copies/mL)であった。
第48週の血中HIV RNA量が400copies/mL未満であった患者の比率は、LPV/r群75%、NFV群62%であった。
血中HIV RNA量が50copies/mL未満であった患者の比率はLPV/r群67%、NFV群52%であった。CD4リンパ球数は、開始時に比べ、LPV/r群で207cells/mm3、NFV群で195cells/mm3増加した。
48週までの治療反応の経過は次図の通り。
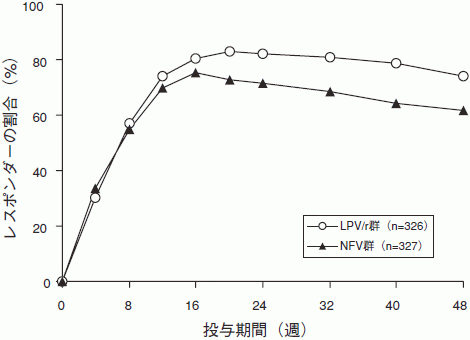
48週までの治療反応の内訳と中止理由は次の通り。
| 結果 | LPV/r+ n=326 |
NFV+ n=327 |
|
|---|---|---|---|
| レスポンダー1 | 75% | 62% | |
| ウイルス学的失敗 | 9% | 25% | |
| (ウイルスリバウンド) | (7%) | (15%) | |
| (VL<400copies/mLに抑制不能) | (2%) | (9%) | |
| 死亡 | 2% | 1% | |
| 有害事象による中止 | 4% | 4% | |
| その他の理由による中止2 | 10% | 8% | |
- 血中HIV RNA(VL)<400copies/mL
- 追跡不能、脱落、コンプライアンス不良、プロトコール違反等
主な有害事象はLPV/r群(326例)で下痢180例(55.2%)、悪心80例(24.5%)、咽頭炎58例(17.8%)、無力症50例(15.3%)等であった。
17.1.2 海外第I/II相試験
試験720(カプセル剤):HIV感染症患者100例を対象として、ロピナビル・リトナビル(LPV/r)の各用量群(第I群:200mg・100mg BID※及び400mg・100mg BID、第II群:400mg・100mg BID及び400mg・200mg BID※)に割り付け、ラミブジン(150mg BID)とサニルブジン(40mg BID)を併用する多施設二重盲検試験を実施した。48~72週が経過した時点で、患者はすべてLPV/r 400mg・100mg BIDのオープンラベル臨床試験に移行した。試験開始時の平均CD4リンパ球数は338cells/mm3(3~918cells/mm3)、平均血中HIV RNA量は4.9log10copies/mL(3.3~6.3log10copies/mL)であった。
360週間後、血中HIV RNA量が400(50)copies/mL未満であった患者は、61%(59%)であり、CD4リンパ球数は501 cells//mm3増加した。360週間の投与期間中、39例が脱落し、このうち15例(15%)は有害事象による中止、1例(1%)は死亡による中止であった。
LPV/r 400mg・100mg BID投与例(51例)における主な有害事象は、下痢29例(56.9%)、異常便27例(52.9%)、頭痛、咳嗽増加各21例(41.2%)、であった6)。
※本剤の承認最大用量は400mg・100mgを1日2回投与である。
17.1.3 海外第I/II相試験
試験765(カプセル剤):HIVプロテアーゼ阻害薬を1剤使用した経験があるが非ヌクレオシド系逆転写酵素阻害薬(NNRTI)治療未経験のHIV感染症患者70名を対象として、ロピナビル・リトナビル(LPV/r)の各用量群(400mg・100mg BID、400mg・200mg BID※)に割り付け、ネビラピン(200mg BID)と2剤のヌクレオシド系逆転写酵素阻害薬(NRTI)を併用する多施設二重盲検試験を実施した。試験開始時における平均CD4リンパ球数は372cells/mm3(72~807cells/mm3)、平均血中HIV RNA量は4.0log10copies/mL(2.9~5.8log10copies/mL)であった。
144週後、血中HIV RNA量が400(50)copies/mL未満であった患者は、54%(50%)であり、CD4リンパ球数は両群平均で212cells/mm3増加した。144週間の投与期間中、27例(39%)が脱落し、このうち、9例(13%)は有害事象による中止、2例(3%)は死亡による中止例であった。
主な有害事象は400/100mg BID群(36例)で下痢21例(58.3%)、悪心14例(38.9%)、咳嗽増加13例(36.1%)、疼痛、嘔吐各12例(33.3%)等であった。
※本剤の承認最大用量は400mg・100mgを1日2回投与である。
17.1.4 海外第I/II相試験
試験940(液剤):出生後6ヵ月以上12歳以下のHIV感染症小児100例、抗HIV化学療法未経験者44例、経験者56例、(共にNNRTIの使用経験なし)を、ロピナビル・リトナビル(LPV/r)230mg//m3・57.5mg//m3 BID及び300mg//m3・75mg//m3 BIDの2群にわけ、逆転写酵素阻害薬(未経験群はサニルブジンとラミブジンを併用し、経験群はネビラピンに加え2剤までのヌクレオシド系逆転写酵素阻害薬)を併用投与する多施設共同オープンラベル並行群間比較試験を実施した。試験開始時における平均CD4リンパ球数は838 cells//mm3で平均血中HIV RNA量は4.7 log10
48週後、未経験群で80%、経験群で71%の患者で血中HIV RNA量が400 copies/mL未満に減少した。CD4リンパ球数は未経験群で平均して404 cells//mm3、経験群で284 cells//mm3増加した。48週の投与期間中2例が脱落した。この試験結果により、6ヵ月以上12歳以下の小児では、ネビラピンを併用しない場合はLPV/r230mg//m3・57.5mg//m3 BID、ネビラピンを併用する場合はLPV/r 300mg//m3・57.5mg//m3 BIDが成人におけるLPV/r 400・100mg BIDの投与(ネビラピンを併用しない場合)に相当するロピナビル血中濃度を得られると考えられた。安全性評価対象100例のうち、主な有害事象は咽頭炎46例(46.0%)、感染38例(38.0%)、咳嗽増加32例(32.0%)、嘔吐、中耳炎各27例(27.0%)等であった7)。
17.3 その他
17.3.1 心電図に対する影響
健康成人39例に本剤400mg・100mg BID及び800mg・200mg BID※を3日間(4回)投与したときのQTcF間隔変化の最大平均値(及び95%上限信頼限界値)はそれぞれ3.6(6.3)msec及び13.1(15.8)msecであった。QTcF間隔がベースラインから60msec以上変化したか500msecを超えた例はなかった。また、3日目において軽度のPR間隔延長が認められた。最大PR間隔は286msecであった(外国人データ)。[9.1.2、10.2参照]
※本剤の承認最大用量は400mg・100mgを1日2回投与である。
18. 薬効薬理
18.1 作用機序
本剤はロピナビルとリトナビルの配合剤である。ロピナビルはHIVプロテアーゼの活性を阻害し、HIVプロテアーゼによるgag-polポリ蛋白質の開裂を抑制することで、感染性を持つ成熟したHIVの産生を抑制する。リトナビルは、CYP3Aによるロピナビルの代謝を競合的に阻害し、ロピナビルの血中濃度の上昇をもたらす。
本剤の抗ウイルス活性は、ロピナビルによるものである。
本剤はHIVプロテアーゼに対する選択的親和性を有し、ヒトのアスパルティックプロテアーゼに対してはほとんど阻害作用を示さない。
18.2 抗ウイルス作用
HIV標準株による感染後早期のリンパ芽球細胞株及び臨床分離株に感染した末梢血リンパ球細胞におけるロピナビルの抗ウイルス作用を検討した。ヒト血清非存在下では、5種類のHIV-1標準株に対するロピナビルの平均EC50は10~27nM(0.006~0.017μg/mL)であり8)、6種類のHIV-1臨床分離株に対するロピナビルの平均EC50は4~11nM(0.003~0.007μg/mL)であった。50%ヒト血清存在下ではHIV-1標準株に対するロピナビルの平均EC50は65~289nM(0.04~0.18μg/mL)であり、7~11倍の効力低下がみられた(in vitro)。
18.3 薬剤耐性
ロピナビルに対する感受性が低下したHIV-1変異株を分離し、ロピナビル単独、あるいは臨床投与時の血中濃度でのロピナビルとリトナビルの存在下にHIV-1のin vitro継代培養を行った。継代培養で分離された株の表現型と遺伝子型を検討したところ、リトナビルの存在はロピナビル耐性株の出現に影響を及ぼさないことが示唆された(in vitro)。
18.3.1 交差耐性
HIVプロテアーゼ阻害薬(PI)間で観察される交差耐性は多様であった。本剤の治療によってロピナビルに対する感受性が低下したウイルスの交差耐性に関する情報はほとんど得られていない。
ロピナビルに対する表現型耐性の増加を認めたPI使用歴のある4例から得られた分離株は本剤投与前からリトナビル、インジナビル、ネルフィナビルに対する交差耐性が維持されていたか、本剤投与後に交差耐性を獲得した。リバウンドしたすべてのウイルスはアンプレナビルに対する感受性を十分に維持していたか、弱い感受性の低下が認められたにとどまった(ロピナビルの最大99倍と比較し、アンプレナビルでは最大8.5倍)。ウイルスのリバウンドを経験した被験者のうち、サキナビルの使用経験のない被験者由来の2株は、サキナビルに対する感受性を維持していた。
18.3.2 ロピナビル・リトナビルを含む併用療法を開始した抗レトロウイルス療法経験患者における抗ウイルス作用減少と遺伝子型との関連
HIVプロテアーゼにアミノ酸置換(L10F/I/R/V、K20M/N/R、L24I、L33F、M36I、I47V、G48V、I54L/T/V、V82A/C/F/S/T、I84V)が3以上存在すると本剤のウイルス学的反応に影響を及ぼすことがわかっている。複数の本剤臨床試験におけるHIVプロテアーゼ阻害薬(PI)耐性変異数と併用療法におけるウイルス学的反応との関係は以下の通りであった。
| PI耐性変異数1 (試験開始時) |
ウイルス学的反応 (48週時) |
||
|---|---|---|---|
| 試験8889) (1種類の n=130 |
試験765 (1種類の n=56 |
試験95710) (複数の n=50 |
|
| 0~2 | 76/103(74%) | 34/45(76%) | 19/20(95%) |
| 3~5 | 13/26(50%) | 8/11(73%) | 18/26(69%) |
| 6以上 | 0/1(0%) | N/A | 1/4(25%) |
- 本分析で検討した置換には、L10F/I/R/V、K20M/N/R、L24I、L33F、M36I、I47V、G48V、I54L/T/V、V82A/C/F/S/T、I84Vが含まれる
- IDV43%、NFV42%、RTV10%、SQV15%
- IDV41%、NFV38%、RTV4%、SQV16%
- IDV86%、NFV54%、RTV80%、SQV70%
18.3.3 HIVプロテアーゼ阻害薬(PI)既使用例における抗ウイルス作用
ロピナビルに対するin vitro感受性低下の臨床的意義を検討するため、複数のPIによる治療にもかかわらず血中HIV RNA量が1,000copies/mLを超えた患者56名に対し本剤を投与し、ウイルスの遺伝子型と表現型を評価した。開始時に分離した56株に対するロピナビルのEC50は、野性株に対するEC50の0.5~96倍であった。48週間にわたり本剤、エファビレンツ及びヌクレオシド系逆転写酵素阻害薬を投与した後、血中HIV RNA量が400copies/mL以下となった患者は、開始時ロピナビル感受性が10倍以下、10倍超~40倍未満、及び40倍以上の患者群でそれぞれ93%(25/27)、73%(11/15)、25%(2/8)であった。また、これら開始時ロピナビル感受性患者群で血中のHIV RNA量が50copies/mL以下となった患者は、それぞれ81%(22/27)、60%(9/15)、25%(2/8)であった。
18.3.4 本剤投与中の耐性ウイルスの選択
227例の抗レトロウイルス療法未経験者及びHIVプロテアーゼ阻害薬(PI)既使用例を対象にした第II相臨床試験では、12~100週間にわたり本剤を服用した後にウイルス量が定量可能(>400copies/mL)であった患者のうち4例の分離株は、試験開始時の分離株に比べ、ロピナビルに対する感受性が著しく低下していた。試験開始時におけるこれら患者4例すべての分離株には、PI耐性に関連する変異が少なくとも4箇所認められた。また、ウイルスリバウンド後では、全ての分離株で変異数が増加しており、PI耐性に関連する変異も含まれていた。しかし、現時点ではデータが不十分なため本剤投与患者における変異パターンがロピナビルによるものかどうかは同定できていない。
19. 有効成分に関する理化学的知見
| 一般的名称 | ロピナビル(Lopinavir)[JAN] |
|---|---|
| 化学名 | (−)-(2S)-N-{(1S,3S,4S)-1-benzyl-4-[2-(2,6-dimethylphenoxy)acetylamino]-3-hydroxy-5-phenylpentyl}-3-methyl-2-(2-oxotetrahydropyrimidin-1-yl)butyramide |
| 分子式 | C37H48N4O5 |
| 分子量 | 628.80 |
| 性状 | 白色~淡黄褐色の粉末で、柔らかい塊を含むこともある。 |
| 構造式 | 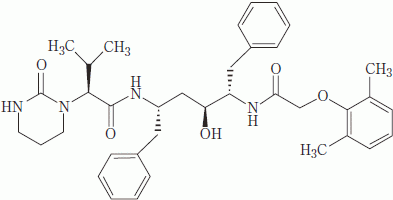 |
| 一般的名称 | リトナビル(Ritonavir)[JAN] |
|---|---|
| 化学名 | (+)-5-thiazolylmethyl[(αS)-α-[(1S,3S)-1-hydroxy-3-[(2S)-2-[3-[(2-isopropyl-4-thiazolyl)methyl]-3-methylureido]-3-methylbutyramido]-4-phenylbutyl]phenethyl]carbamate |
| 分子式 | C37H48N6O5S2 |
| 分子量 | 720.94 |
| 性状 | 白色~淡黄褐色の粉末で、柔らかい塊を含むこともある。< |
| 構造式 | 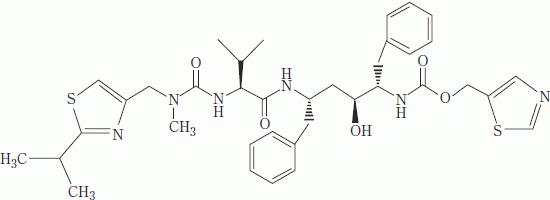 |
22. 包装
160mL[1瓶]
23. 主要文献
- Zha, J., et al.:Pharmacol Res Perspect. 2022;10(6):e01024
- Molla,A.,et al., Virology., 250, 255-262,
- Kumar,GN.,et al., Drug Metab.Dispos., 27, 86-91
- Kumar,GN.,et al., Drug Metab.Dispos., 27, 902-908
- Peng,JZ.,et al., J.Clin.Pharmacol., 46, 265-274
- Murphy,RL.,et al., HIV Clin.Trials., 9, 1-10
- Saez-Llorens,X.,et al., Pediatr.Infect.Dis.J., 22, 216-223
- Sham,HL.,et al., Antimicrob.Agents Chemother., 42, 3218-3224
- Pollard,RB.,et al., 7thInt.Cong.Drug Ther.HIV.2004:7:Abstract PL 3.2
- Kempf,DJ.,et al., Antivir.Ther., 7, 165-174
24. 文献請求先及び問い合わせ先
アッヴィ合同会社 くすり相談室
〒108-0023 東京都港区芝浦3-1-21
フリーダイヤル 0120-587-874
26. 製造販売業者等
26.1 製造販売元
アッヴィ合同会社
東京都港区芝浦3-1-21