シュンレンカ皮下注463.5mgの添付文書
- 商品名:
- シュンレンカ皮下注463.5mg
- 一般名:
- レナカパビルナトリウム皮下注
- 略称 :
- LEN

添付文書の読み方
ここで提供している添付文書情報は、2023年10月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
抗ウイルス化学療法剤(HIVカプシド阻害剤)
レナカパビルナトリウム皮下注
- *2025年3月改訂(第2版)
- 2023年8月作成(第1版)
規制区分:処方箋医薬品注)
注)注意-医師等の処方箋により使用すること
| 日本標準商品分類番号 | 87625 |
|---|
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 36ヵ月 |
| 承認番号 | 30500AMX00183000 |
|---|---|
| 販売開始 | 2023年9月 |
2. 禁忌(次の患者には投与しないこと)
2.1
本剤の成分に対し過敏症の既往歴のある患者
2.2
リファンピシン、フェニトイン、フェニトイン・フェノバルビタール、ホスフェニトインナトリウム水和物、カルバマゼピン、アパルタミド、エンザルタミド、ミトタン、セイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品、ロミタピドメシル酸塩、メチルエルゴメトリンマレイン酸塩及びエルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリンを投与中の患者[10.1、16.7.2 参照]
3. 組成・性状
3.1 組成
| 有効成分・含量 (1バイアル中)注) |
1.5mL中にレナカパビルナトリウム473.1mg (レナカパビルとして463.5mg) |
|---|---|
| 添加剤 (1バイアル中)注) |
マクロゴール300 896.3mg |
3.2 製剤の性状
| 色・剤形 | 黄色澄明の液で、たやすく検出される不溶性 異物を認めない |
|---|---|
| pH(参考値) | 9.0-10.2 |
4. 効能又は効果
多剤耐性HIV-1感染症
5. 効能又は効果に関連する注意
以下のいずれも満たす患者に投与すること。[17.1.1 参照]
- 過去の治療において、本剤を含まない既存の抗レトロウイルス療法による適切な治療を行ってもウイルス学的抑制が得られなかった患者
- 薬剤耐性検査(遺伝子型解析あるいは表現型解析)を実施し、本剤を含まない複数の抗HIV薬に耐性を示す患者
- 本剤の投与の前にレナカパビル経口剤を投与し、レナカパビルに対する忍容性が確認された患者
6. 用法及び用量
通常、成人にはレナカパビル経口剤の投与開始後15日目に、レナカパビルとして927mgを皮下投与する。以降は、927mgを6ヵ月に1回、皮下投与する。投与に際しては、必ず他の抗HIV薬と併用すること。
7. 用法及び用量に関連する注意
7.1
併用する抗HIV薬は、患者の治療歴及び薬剤耐性検査(遺伝子型解析あるいは表現型解析)を参考に選択すること。
7.2
本剤の投与スケジュールを遵守すること。投与スケジュールを遵守できなかった場合は、本剤の継続の可否も含め、治療法を再考すること。
7.3
本剤の2回目以降の投与は、投与予定日(本剤の最終投与日から26週間)の前後2週間以内に行うこと。
7.4
本剤の最終投与日から28週間超経過したが、本剤投与を再開することが医療上適切である場合、レナカパビル経口剤の投与1日目から再開すること。レナカパビル経口剤を再開する際にはレナカパビル経口剤の電子添文を参照すること。
8. 重要な基本的注意
8.1
本剤による治療は、抗HIV療法に十分な経験を持つ医師のもとで開始すること。
8.2
本剤は投与スケジュールが遵守されない場合、ウイルスの再増殖及び薬剤耐性リスクのおそれがあるため、投与スケジュールを遵守するよう患者に指導すること。
8.3
本剤の投与を中止する場合は、以下の点に留意すること。
8.3.1
本剤は投与後に長期間(12ヵ月以上)にわたって血中に残留する可能性があるため、本剤の長期作用に注意すること。[9.5、9.6、10. 参照]
8.3.2
ウイルス耐性の発現リスクを最小限に抑えるため、可能であれば本剤最終投与後28週間以内に、他の抗レトロウイルス療法を開始すること。
8.4
本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又はそれに代わる適切な者に次の事項についてよく説明し同意を得た後、使用すること。
8.4.1
本剤はHIV感染症の根治療法薬ではないことから、日和見感染症を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化についてはすべて担当医に報告すること。
8.4.2
本剤の長期投与による影響については現在のところ不明であること。
8.4.3
本剤は併用薬剤と相互作用を起こすことがあるため、服用中のすべての薬剤を担当医に報告すること。また、本剤で治療中に新たに他の薬剤を服用する場合、事前に担当医に相談すること。[10.、16.7.1、16.7.2 参照]
8.5
本剤を含む抗HIV薬の多剤併用療法を行った患者で、免疫再構築炎症反応症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
9. 特定の背景を有する患者に関する注意
9.2 腎機能障害患者
9.2.1 末期腎不全患者(クレアチニンクリアランスが15mL/min未満)
末期腎不全患者(クレアチニンクリアランス15mL/min未満)を対象とした臨床試験は実施していない。本剤の血漿中濃度が上昇する可能性がある。
9.3 肝機能障害患者
9.3.1 重度の肝機能障害患者(Child-Pugh分類C)
重度の肝機能障害患者(Child-Pugh分類C)を対象とした臨床試験は実施していない。本剤の血漿中濃度が上昇する可能性がある。
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。本剤は投与後に長期間(12ヵ月以上)にわたって血中に残留する可能性があるため、妊娠した場合に胎児が本剤に曝露される可能性がある。動物実験(ラット)で乳汁又は胎盤を介して出生児にレナカパビルが移行した報告がある。[8.3.1 参照]
9.6 授乳婦
授乳を避けさせること。一般に、乳児へのHIV感染を防ぐため、あらゆる状況下においてHIVに感染した女性は授乳をすべきでない。本剤の最後の投与から長期間(12ヵ月以上)にわたって本剤が乳汁中に認められる可能性がある。動物実験(ラット)で乳汁又は胎盤を介して出生児にレナカパビルが移行した報告がある。ヒトにおける乳汁への移行は不明である。[8.3.1 参照]
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能が低下しており、合併症や他の薬剤の併用が多い。
*10. 相互作用
レナカパビルはCYP3A、P-gp及びUGT1A1の基質であり、CYP3Aの中程度の阻害薬及びP-gpの阻害薬である。[8.3.1 参照],[8.4.3 参照]
10.1 併用禁忌(併用しないこと)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
|
レナカパビルの血漿中濃度が低下するため、本剤の効果が減弱し、本剤に対する耐性が発現する可能性がある。 | これら薬剤の強いCYP3A、P-gp又はUGT1A1の誘導作用により、本剤の血漿中濃度が低下する可能性がある。 |
| セイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品 [2.2 参照] |
セント・ジョーンズ・ワートの強いCYP3A、P-gp及びUGT1A1の誘導作用により、本剤の血漿中濃度が低下する可能性がある。 | |
| ロミタピドメシル酸塩 (ジャクスタピッド) [2.2 参照] |
ロミタピドメシル酸塩の血漿中濃度が上昇する可能性がある。 | レナカパビルのCYP3A阻害作用により、ロミタピドメシル酸塩の血漿中濃度が上昇する可能性がある。 |
|
これら薬剤の血漿中濃度が上昇する可能性がある。 | レナカパビルのCYP3A阻害作用により、これら薬剤の血漿中濃度が上昇する可能性がある。 |
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| *ジゴキシン | ジゴキシンの血漿中濃度が上昇する可能性がある。本剤と併用する場合は、ジゴキシンの血漿中濃度のモニタリングを行うこと。 | レナカパビルのP-gp阻害作用により、ジゴキシンの血漿中濃度が上昇する可能性がある。 |
| *直接経口抗凝固薬 (DOAC)
|
これら薬剤の血漿中濃度が上昇する可能性がある。 | レナカパビルのCYP3A又はP-gp阻害作用により、これら薬剤の血漿中濃度が上昇する可能性がある。 |
| シンバスタチン | シンバスタチンの血漿中濃度が上昇する可能性がある。シンバスタチンは最低用量から開始し、安全性(ミオパチーなど)をモニタリングしながら慎重に増量すること。 | レナカパビルのCYP3A阻害作用により、シンバスタチンの血漿中濃度が上昇する可能性がある。 |
| *コルチコステロイド (全身性)
|
全身性コルチコステロイドの曝露量が著しく上昇する可能性がある。これら薬剤は最低用量から開始し、安全性をモニタリングしながら慎重に増量すること。 また、全身性デキサメタゾンとの併用によりレナカパビルの血漿中濃度が低下し、特に長期間投与する場合は、本剤の効果が減弱し、本剤に対する耐性が発現する可能性があるため、他のコルチコステロイドへの代替を検討すること。 |
レナカパビルのCYP3A阻害作用により、コルチコステロイドの曝露量が著しく上昇し、クッシング症候群及び副腎抑制のリスクが増加する可能性がある。 また、デキサメタゾンのCYP3A誘導作用により、レナカパビルの血漿中濃度が低下する可能性がある。 |
|
これら薬剤の血漿中濃度が上昇する可能性がある。 | レナカパビルのCYP3A阻害作用により、これら薬剤の血漿中濃度が上昇する可能性がある。 |
ホスホジエステラーゼ5(PDE-5)阻害薬
|
これら薬剤の血漿中濃度が上昇する可能性がある。勃起不全の治療のためにこれら薬剤を本剤と併用する場合は、これら薬剤は最低用量から開始すること。肺動脈性肺高血圧症の治療のためにタダラフィルを本剤と併用することは推奨されない。 | レナカパビルのCYP3A阻害作用により、これら薬剤の血漿中濃度が上昇する可能性がある。 |
| アタザナビル/リトナビル | レナカパビルの血漿中濃度が上昇する可能性がある。アタザナビル/リトナビルと本剤の併用は推奨されない。 | アタザナビル/リトナビルの強いCYP3A、P-gp及びUGT1A1阻害作用により、レナカパビルの血漿中濃度が上昇する可能性がある。 |
| エファビレンツ [16.7.2 参照] |
レナカパビルの血漿中濃度が低下する可能性があり、本剤の効果が減弱し、本剤に対する耐性が発現する可能性がある。エファビレンツと本剤の併用は推奨されない。 | エファビレンツのCYP3A、P-gp及びUGT1A1誘導作用により、レナカパビルの血漿中濃度が低下した。 |
|
レナカパビルの血漿中濃度が低下する可能性があり、本剤の効果が減弱し、本剤に対する耐性が発現する可能性がある。これら薬剤と本剤の併用は推奨されない。 | これら薬剤の中程度のCYP3A、P-gp又はUGT1A1誘導作用により、レナカパビルの血漿中濃度が低下する可能性がある。 |
11. 副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.2 その他の副作用
11.2 その他の副作用
| 器官分類 | 3%以上 |
|---|---|
| 胃腸障害 | 悪心 |
| 一般・全身障害および投与部位の状態 | 注射部位反応(腫脹、疼痛、結節、紅斑、硬結、そう痒感、漏出、不快感、腫瘤、血腫、浮腫、潰瘍)(63%) |
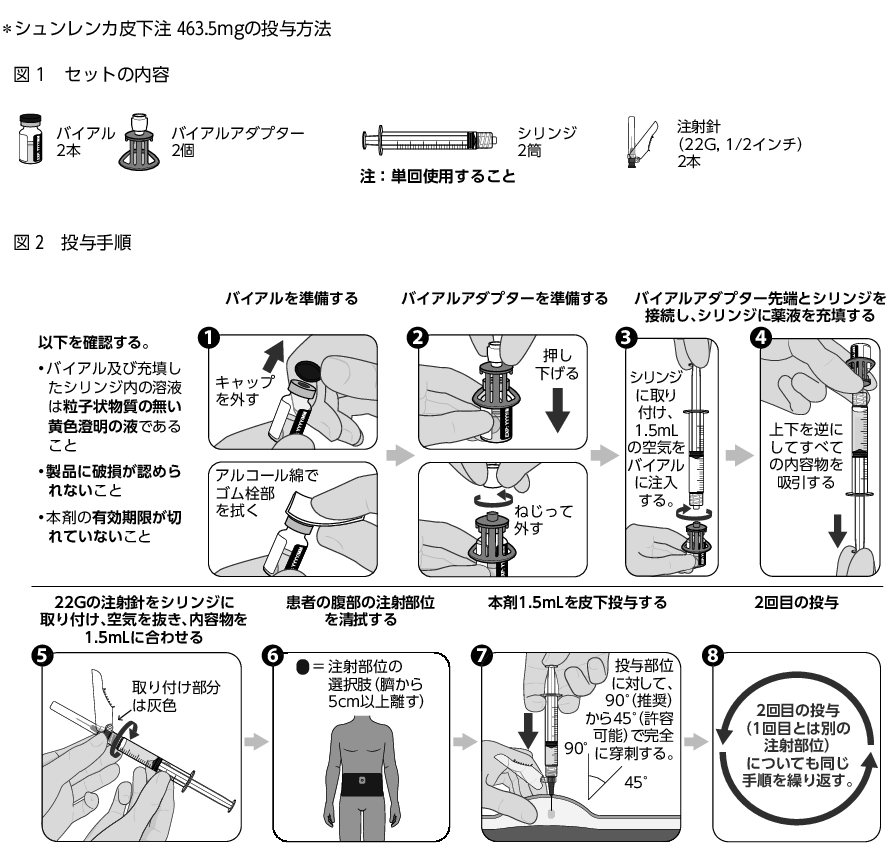
14. 適用上の注意
14.1 薬剤投与前の注意
14.1.1
投与前にバイアル内の溶液に粒子状物質及び変色がないか目視で確認すること。粒子状物質が含まれていたり溶液が変色している場合は、使用しないこと。
14.1.2
溶液をバイアルから取り出すときは、添付のバイアルアダプターを用いること。
14.1.3
溶液をバイアルから取り出したら、速やかに使用すること。
14.1.4
各バイアルは1回限りの使用とし、残液は廃棄すること。
14.2 薬剤投与時の注意
*14.2.1
注射部位1カ所あたり1.5mLを皮下に投与し、皮内には投与しないこと。皮内投与により、壊死や潰瘍などの重篤な注射部位反応を起こすことがある。
14.2.2
注射部位は、腹部とし、臍から5cm以上離すこと。
14.2.3
同一箇所への2本の注射は避け、投与ごとに注射部位を変えること。
14.2.4
皮膚が敏感な部位、皮膚に異常のある部位(傷、発疹、硬結等)には注射しないこと。
14.2.5
バイアルアダプター、シリンジ、注射針は再使用しないこと。
16. 薬物動態
16.1 血中濃度
16.1.1 健康被験者
外国人健康被験者にレナカパビル927mgを単回皮下投与したときのレナカパビルの薬物動態パラメータは下表のとおりであった。
| 薬物動態パラメータ | 927mg(1.5mL×2) (8例) |
|---|---|
| AUCinf(h・ng/mL) | 232382.4(28.7)b |
| Cmax(ng/mL) | 61.2(43.5) |
| Tmax(day)a | 84.0 |
| t1/2(day)a | 80.8b |
| CL/F(L/h) | 4.2(29.0)b |
*16.1.2 HIV-1感染者
多剤治療歴を有するHIV-1感染者にレナカパビルを経口及び皮下投与したときの母集団薬物動態パラメータの推定値は下表のとおりであった。1)
| 薬物動態 パラメータ |
投与初日及び2日目:600mg経口投与、 8日目:300mg経口投与、 15日目:927mg皮下投与 |
|
|---|---|---|
| 投与初日~15日目 | 投与15日目~6ヵ月 | |
| AUCtau (h・ng/mL) |
25962.9(67.8) | 251907.2(48.2) |
| Cmax (ng/mL) |
124.4(85.1) | 87.3(49.4) |
| Ctrough (ng/mL) |
48.6(52.1) | 35.1(59.2) |
母集団薬物動態解析に基づくと、多剤治療歴のあるHIV-1感染者におけるレナカパビルの曝露量(AUCtau、Cmax及びCtrough)はHIV-1非感染者よりも29%~84%高かった。1)
16.2 吸収
16.2.1 バイオアベイラビリティ
皮下投与後のレナカパビルは完全に吸収される。2)
*16.3 分布
レナカパビルの血漿蛋白結合率は約99.8%であった(ex vivoデータ)。3)
母集団薬物動態解析に基づくと、多剤治療歴のあるHIV-1感染者におけるレナカパビルの定常状態の分布容積は976Lであった。
16.4 代謝
外国人健康被験者に14C標識レナカパビル20mgを単回静脈内投与注)したとき、血漿中には主に未変化体(血漿中総放射能の69%)が検出され、血漿中総放射能の10%を超える代謝物は検出されなかった。レナカパビルの消失における代謝の寄与は小さい。レナカパビルは、主にCYP3A及びUGT1A1を介する酸化、N-脱アルキル化、水素化、アミド加水分解、グルクロン酸抱合、ヘキソース抱合、ペントース抱合及びグルタチオン抱合により代謝された。4)
16.5 排泄
外国人健康被験者に14C標識レナカパビル20mgを単回静脈内投与注)したとき、投与した放射能の76%が糞中に排泄され、尿中への排泄は1%未満であった。糞中には主に未変化体(投与量の33%)が検出された。4)
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
重度の腎機能障害を有する被験者(クレアチニンクリアランスが15mL/min以上30mL/min未満)にレナカパビル300mgを単回経口投与注)したとき、レナカパビルのAUCinf及びCmaxは、腎機能正常被験者と比べて、それぞれ84%及び162%増加した。5)(外国人のデータ)
16.6.2 肝機能障害患者
中等度の肝機能障害を有する被験者(Child-Pugh分類B)にレナカパビル300mgを単回経口投与注)したとき、レナカパビルのAUCinf及びCmaxは、肝機能正常被験者と比べて、それぞれ47%及び161%増加した。6)(外国人のデータ)
16.7 薬物相互作用
16.7.1 非臨床における薬物相互作用試験
レナカパビルはCYP3A及びOATP1B1に対して阻害作用を示す。[8.4.3参照]
16.7.2 臨床における薬物相互作用試験
薬物相互作用試験の結果は下表のとおりであった。7)(外国人のデータ)[2.2、8.4.3、10.1、10.2 参照]
| 併用薬 | 併用薬の用量 | 例数 | 併用時/単独投与時のレナカパビルの 薬物動態パラメータ比 (90%信頼区間) |
|
|---|---|---|---|---|
| Cmax | AUC | |||
| コビシスタット (食後) |
150mg 1日1回 | 29 | 2.10 (1.62, 2.72) |
2.28 (1.75, 2.96) |
| ダルナビル/コビシスタット (食後) |
800mg/150mg 1日1回 | 29 | 2.30 (1.79, 2.95) |
1.94 (1.50, 2.52) |
| ボリコナゾール (空腹時) |
400mg 1日2回、 200mg 1日2回c |
25 | 1.09 (0.81, 1.47) |
1.41 (1.10, 1.81) |
| アタザナビル/コビシスタット (食後)[10.2 参照] |
300mg/150mg 1日1回 | 21 | 6.60 (4.99, 8.73) |
4.21 (3.19, 5.57) |
| リファンピシン (空腹時)[2.2、10.1 参照] |
600mg 1日1回 | 25 | 0.45 (0.34, 0.60) |
0.16 (0.12, 0.20) |
| エファビレンツ (空腹時)[10.2 参照] |
600mg 1日1回 | 18 | 0.64 (0.45, 0.92) |
0.44 (0.32, 0.59) |
| ファモチジン (本剤投与の2時間前/空腹時) |
40mg 1日1回 | 25 | 1.01 (0.75, 1.34) |
1.28 (1.00, 1.63) |
| 併用薬 | 併用薬の用量 | 例数 | 併用時/単独投与時の併用薬の 薬物動態パラメータ比 (90%信頼区間) |
|
|---|---|---|---|---|
| Cmax | AUC | |||
| テノホビル アラフェナミド (食後) |
25mg単回投与 | 28 | 1.24 (0.98, 1.58) |
1.32 (1.09, 1.59) |
| テノホビルc | 1.23 (1.05, 1.44) |
1.47 (1.27, 1.71) |
||
| ピタバスタチン (本剤と同時投与/食後) |
2mg単回投与 | 30 | 1.00 (0.84, 1.19) |
1.11 (1.00, 1.25) |
| ピタバスタチン (本剤投与の3日後/食後) |
2mg単回投与 | 28 | 0.85 (0.69, 1.05) |
0.96 (0.87, 1.07) |
| ロスバスタチン (食後) |
5mg単回投与 | 30 | 1.57 (1.38, 1.80) |
1.31 (1.19, 1.43) |
| ミダゾラム (本剤と同時投与/食後)[10.2 参照] |
2.5mg単回投与 | 28 | 1.94 (1.81, 2.08) |
3.59 (3.30, 3.91) |
| 1-hydroxymidazolamd[10.2 参照] | 0.54 (0.50, 0.59) |
0.76 (0.72, 0.80) |
||
| ミダゾラム(本剤投与の1日後/食後)[10.2 参照] | 2.5mg単回投与 | 28 | 2.16 (2.02, 2.30) |
4.08 (3.77, 4.41) |
| 1-hydroxymidazolamd[10.2 参照] | 0.52 (0.48, 0.57) |
0.84 (0.80, 0.88) |
||
*17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第II/III相臨床試験
多剤治療歴を有する多剤耐性HIV-1感染者を対象とし、レナカパビルナトリウムの有効性及び安全性を検討することを目的として、無作為化部分盲検プラセボ対照並行群間比較試験(GS-US-200-4625試験)を実施した。8)主な選択基準は表1のとおりであった。
| 選択基準 |
|
|---|
コホート1の14日間の並行群間比較期では、治療不成功となったレジメンの投与を継続するとともに投与開始1及び2日目にレナカパビル経口剤600mg又はプラセボ、8日目にレナカパビル経口剤300mg又はプラセボを投与した。並行群間比較期以降は、レナカパビル群の被験者は投与開始15日目より最適なバックグラウンドレジメン(OBR)とともにレナカパビル注射剤927mgを26週間に1回皮下継続投与し、プラセボ群の被験者はOBRとともに投与開始15及び16日目にレナカパビル経口剤600mg、22日目にレナカパビル経口剤300mg、29日目よりレナカパビル注射剤927mgを26週間に1回皮下投与した。コホート2では、OBRとともに投与開始1及び2日目にレナカパビル経口剤600mg、8日目にレナカパビル経口剤300mg、15日目よりレナカパビル注射剤927mgを26週間に1回皮下投与した。
主要有効性評価項目は、コホート1において14日間の並行群間比較期終了時のHIV-1 RNA量がベースラインから0.5log10copies/mL以上減少した被験者の割合とされ、結果は表2のとおりであり、プラセボに対するレナカパビルの優越性が検証された。
| レナカパビル群 | プラセボ群 | |
|---|---|---|
| HIV-1 RNA量が0.5log10copies/mL以上減少した被験者の割合 | 87.5% (21/24例) |
16.7% (2/12例) |
| 群間差(95%信頼区間) | 70.8%(34.9, 90.0) | |
| p値 | <0.0001 | |
コホート1における26週及び52週時のウイルス学的抑制が得られた被験者の割合は表3のとおりであった。
| レナカパビルとOBR併用 | ||
|---|---|---|
| 26週 | 52週 | |
| HIV-1 RNA量50copies/mL未満 | 81%(29/36) | 83%(30/36) |
コホート1及びコホート2においてレナカパビルを投与された72例中50例(69.4%、最終被験者がレナカパビル注射剤の皮下投与開始から52週経過時の解析)に副作用が認められた。主な副作用は、注射部位腫脹26例(36.1%)、注射部位疼痛22例(30.6%)、注射部位紅斑22例(30.6%)及び注射部位結節18例(25.0%)であった。
[5. 参照]
18. 薬効薬理
18.1 作用機序
レナカパビルは、HIV-1のカプシドタンパク単量体間の界面に直接結合しHIV-1プロウイルスDNAのカプシド介在性核内取込み、ウイルスの形成及び放出並びにカプシドコア形成等のウイルス複製における複数の重要な段階に関与するHIV-1カプシドタンパクの機能を阻害することにより、HIV-1の複製を阻害する。9)
18.2 抗ウイルス活性
HIV-1の実験室株及び臨床分離株に対するレナカパビルの抗ウイルス活性をMT-4細胞(リンパ芽球様細胞株)、末梢血単核球、初代培養単球/マクロファージ細胞及びCD4陽性Tリンパ球を用いて評価し、野生型HIV-1ウイルスに対するEC50は、0.03~0.19nmol/Lであった。10)レナカパビルのタンパク質補正EC95は、野生型HIV-1ウイルスのMT-4 T細胞株で4nmol/L(3.87ng/mL)であった。10)
レナカパビルと主要なクラスの抗レトロウイルス薬(NRTI、NNRTI、INSTI、PI)の代表的薬剤を併用した試験において、相乗的な抗ウイルス効果が認められた。これらの併用では拮抗作用は認められなかった。11)レナカパビルは、培養細胞系においてHIV-1のすべてのグループM、N及びO(サブタイプA、A1、CRF01_AE、CRF01_AG、B、CRF12_BF、C、D、E、F、G、H)に対して抗ウイルス活性を示した。12)
18.3 薬剤耐性
18.3.1 In vitro試験
レナカパビルを用いたin vitro耐性選択試験により、レナカパビルに対する感受性の低下を示したカプシドタンパクの7つの変異(L56I、M66I、Q67H、K70N、N74D/S及びT107Nの単一又は二重変異)が同定され、当該変異導入株におけるレナカパビルに対する感受性は、野生型ウイルスに比べて4倍から3,226倍超低下した。レナカパビルに対する感受性が野生型ウイルスの10倍超低下した変異導入株では、初代ヒトCD4陽性Tリンパ球及びマクロファージにおける複製能の低下が認められた(野生型ウイルス量のそれぞれ0.03~28%及び1.9~72%)。13)
*18.3.2 臨床試験
GS-US-200-4625試験では、31%(22/72例)の被験者が52週時の耐性解析の基準(ウイルス学的失敗の確定時点でHIV-1 RNA量が50copies/mL以上[4週時点のウイルス学的効果不十分、最終来院時のウイルス学的リバウンド又はウイルス血症])を満たしたことから、レナカパビル耐性に関連するカプシドタンパク変異が解析された。レナカパビル耐性に関連するカプシドタンパク変異は、13%(9例)に認められた。被験者の8.3%(6例)にM66I変異が認められ、M66I単一又は他の変異(Q67H/K/N、K70N/R/S、N74D/H、A105T及び/又はT107A/C)との組み合わせであった。M66I変異が認められなかった3例で、Q67H/K、K70H/R、A105S/T及びT107Nが認められた。
表現型分析において、M66I変異、K70H+A105A/S/T+T107T/N変異、Q67K+K70H変異、Q67H+K70R変異、Q67Q/H変異を有する株におけるレナカパビルに対する感受性は、野生型ウイルスと比較してそれぞれ234倍(中央値)、265倍、342倍、15倍及び5.9倍低下した。8),15)
18.4 交差耐性
主要なクラスの抗レトロウイルス薬(NRTI、NNRTI、INSTI、PI)に耐性を示すHIV-1部位特異的変異株及び患者由来のHIV-1臨床分離株(58例)、並びに成熟阻害薬に耐性を示すHIV-1臨床分離株(32例)及び侵入阻害薬(EI)クラス(Fostemsavir、Ibalizumab、マラビロク及びEnfuvirtide)に耐性を示すHIV-1臨床分離株(42例)に対するレナカパビルのin vitro抗ウイルス活性を測定した。14)
これらすべての変異株に対してレナカパビルの活性の大きな変化は認められなかった。また、レナカパビルの抗ウイルス活性は、自然発生するGag多型の存在による影響を受けなかった。
19. 有効成分に関する理化学的知見
| 一般的名称 | レナカパビルナトリウム Lenacapavir Sodium(JAN) |
|---|---|
| 化学名 | Monosodium[4-chloro-7-{2-[(1S)-1-{2-[(3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1Hcyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl]acetamido}-2-(3,5-difluorophenyl)ethyl]-6-[3-(methanesulfonyl)-3-methylbut-1-yn-1-yl]pyridin-3-yl}-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl](methanesulfonyl)azanide |
| 分子式 | C39H31ClF10N7NaO5S2 |
| 分子量 | 990.26 |
| 化学構造式 | 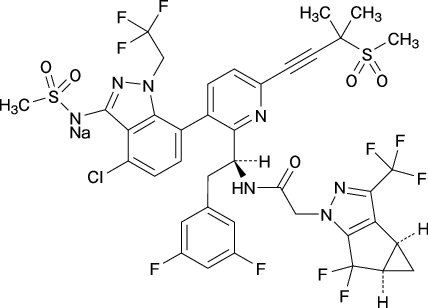 |
| 性状 | 淡黄色~黄色の固体 |
| 溶解性 | N,N-ジメチルホルムアミド、メタノール、N-メチル-2-ピロリドン、マクロゴール300、アセトン又はジメチルスルホキシドに溶けやすく、アセトニトリルにやや溶けにくく、エタノールに溶けにくく、2-プロパノール又はジクロロメタンに極めて溶けにくい。 |
| 融点 | 約228℃ |
| 分配係数 | Log P=5.1(オクタノール/水) |
20. 取扱い上の注意
本剤は遮光する必要があるため、使用直前まで外箱に入れて保存すること。
21. 承認条件
21.1
医薬品リスク管理計画を策定の上、適切に実施すること。
21.2
本剤の使用に当たっては、患者に対して本剤に関して更なる有効性・安全性のデータを引き続き収集中であること等を十分に説明し、インフォームドコンセントを得るよう、医師に要請すること。
21.3
現在実施中又は計画中の臨床試験については、終了後速やかに試験成績及び解析結果を提出すること。
21.4
日本人を対象とした薬物動態試験を実施し、その進捗状況を定期的に報告するとともに、終了後速やかに試験成績及び解析結果を提出すること。
21.5
再審査期間が終了するまでの間、原則として国内の全投与症例を対象とした製造販売後調査を実施し、本剤の使用実態に関する情報(患者背景、有効性・安全性(他剤併用時の有効性・安全性を含む)及び薬物相互作用のデータ等)を収集して定期的に報告するとともに、調査の結果を再審査申請時に提出すること。
22. 包装
2バイアル(シリンジ2筒、注射針(22G、12.7mm)2本、バイアルアダプター2個を添付)
23. 主要文献
- 社内資料:CTRA-2021-1054試験(2023年8月1日承認)
- 社内資料:(2023年8月1日承認、CTD 2.7.2.3.1.1)
- 社内資料:(2023年8月1日承認、CTD 2.7.2.3.1.2)
- 社内資料:GS-US-200-4329試験(2023年8月1日承認、CTD 2.7.2.2.2.1)
- 社内資料:GS-US-200-4330試験(2023年8月1日承認、CTD 2.7.2.2.4.1)
- 社内資料:GS-US-200-4331試験(2023年8月1日承認、CTD 2.7.2.2.4.2)
- 社内資料:GS-US-200-4333試験(2023年8月1日承認、CTD 2.7.2.2.5.1, 2.7.2.3.1.7)
- 社内資料:GS-US-200-4625試験(2023年8月1日承認、CTD 2.7.2.4.2.1, 2.7.3.2, 2.7.3.3, 2.7.4.1, 2.7.4.2)、Sorana SegalMaurer, et. al:N Engl J Med. 2022;386:1793-803
- 社内資料:PC-200-2022試験、PC-200-2023試験、PC-200-2024試験、PC-200-2036試験、PC-200-2037試験(2023年8月1日承認、CTD 2.7.2.4.1.1)
- 社内資料:AD-200-2020試験(2023年8月1日承認、CTD 2.6.2.2.1.1)
- 社内資料:PC-200-2030試験(2023年8月1日承認、CTD 2.6.2.5.1)
- 社内資料:PC-200-2020試験、PC-200-2041試験(2023年8月1日承認、CTD 2.6.2.2.1.3, 2.6.2.2.1.4)
- 社内資料:PC-200-2026試験(2023年8月1日承認、CTD 2.6.2.2.4.2, 2.6.2.2.4.5)
- 社内資料:PC-200-2027試験、PC-200-2044試験、PC-200-2037試験、PC-200-2043試験(2023年8月1日承認、CTD 2.6.2.2.3, 2.7.2.4.2.1)
- 社内資料:PC-200-2051試験
24. 文献請求先及び問い合わせ先
ギリアド・サイエンシズ株式会社
メディカルサポートセンター
〒100-6616 東京都千代田区丸の内一丁目9番2号
グラントウキョウサウスタワー
フリーダイアル 0120-506-295
FAX 03-5958-2959
受付時間:9:00~17:30(土・日・祝日及び会社休日を除く)
26. 製造販売業者等
26.1 製造販売元
ギリアド・サイエンシズ株式会社
東京都千代田区丸の内1-9-2
グラントウキョウサウスタワー