レクシヴァ錠700の添付文書
- 商品名:
- レクシヴァ錠700
- 一般名:
- ホスアンプレナビル
- 略称 :
- FPV

添付文書の読み方
ここで提供している添付文書情報は、2025年2月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
抗ウイルス化学療法剤
ホスアンプレナビルカルシウム水和物錠
- **2024年8月改訂(第5版)
*22023年8月改訂(第4版) - 規制区分:劇薬、処方箋医薬品注)
注)注意-医師等の処方箋により使用すること
| 日本標準商品分類番号 | 87625 |
|---|
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 3年 |
| 承認番号 | 21600AMZ00652 |
|---|---|
| 販売開始 | 2005年1月 |
2. 禁忌(次の患者には投与しないこと)
2.1
本剤の成分あるいはアンプレナビルに対して過敏症の既往歴のある患者
2.2
重度の肝障害患者[9.3.1、16.6.2参照]
2.3
肝代謝酵素CYP3A4で代謝される薬剤で治療域が狭い薬剤(ベプリジル塩酸塩水和物、シサプリド、ピモジド、トリアゾラム、ミダゾラム、エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン、ジヒドロエルゴタミンメシル酸塩、エルゴメトリンマレイン酸塩、メチルエルゴメトリンマレイン酸塩)を投与中の患者[10.1 参照]
2.4
バルデナフィル塩酸塩水和物を投与中の患者[10.1 参照]
2.5
リファンピシンを投与中の患者[10.1 参照],[16.7.3 参照]
3. 組成・性状
3.1 組成
| 販売名 | レクシヴァ錠700 |
|---|---|
| 有効成分 | 1錠中 ホスアンプレナビルカルシウム水和物をホスアンプレナビルとして700mg |
| 添加剤 | 結晶セルロース、クロスカルメロースナトリウム、ポビドン、ステアリン酸マグネシウム、軽質無水ケイ酸、ヒプロメロース、酸化チタン、トリアセチン、三二酸化鉄 |
3.2 製剤の性状
| 販売名 | レクシヴァ錠700 |
|---|---|
| 剤形・性状 | 淡紅白色のフィルムコーティング錠 |
| 識別コード | GXLL7 |
| 表 (長径×短径) |
 20.5mm×9.5mm |
| 裏 |  |
| 側面 (厚さ) |
 7.4mm |
| 質量 | 1174mg |
4. 効能又は効果
HIV感染症
5. 効能又は効果に関連する注意
5.1
無症候性ヒト免疫不全ウイルス(HIV)感染症に関する治療開始については、CD4リンパ球数及び血漿中HIV RNA量が指標とされている。よって、本剤の使用にあたっては、患者のCD4リンパ球数及び血漿中HIV RNA量を確認するとともに、最新のガイドライン1),2),3)を確認すること。
5.2
本剤のHIV-2感染症患者に対する有効性・安全性は確認されていない。
6. 用法及び用量
通常、成人には以下の用法・用量に従い経口投与する。投与に際しては、必ず他の抗HIV薬と併用すること。
〈抗HIV薬の治療経験がない患者〉
- ホスアンプレナビルとして1回700mgとリトナビル1回100mgをそれぞれ1日2回併用投与
- ホスアンプレナビルとして1回1400mgとリトナビル1回100mg又は200mgをそれぞれ1日1回併用投与
- ホスアンプレナビルとして1回1400mgを1日2回投与
〈HIVプロテアーゼ阻害剤の投与経験がある患者〉
- ホスアンプレナビルとして1回700mgとリトナビル1回100mgをそれぞれ1日2回併用投与
7. 用法及び用量に関連する注意
7.1
HIVは感染初期から多種多様な変異株を生じ、薬剤耐性を発現しやすいことが知られているので、他の抗HIV薬と併用すること。
7.2
HIVプロテアーゼ阻害剤投与経験のある患者に対する本剤及びリトナビル1日1回併用投与は推奨されない。
7.3
抗HIV薬の治療経験がない患者でリトナビルの投与が困難な患者に対しては、リトナビルと併用しない用法・用量(ホスアンプレナビルとして1回1400mgを1日2回)の適用を考慮すること。
7.4
ホスアンプレナビルとリトナビルの併用投与において、「用法及び用量」で定められた用量よりも高用量の投与により、AST、ALTが上昇する可能性があるため、「用法及び用量」で定められた用量を超えて投与しないこと。
7.5
軽度又は中等度の肝機能障害患者に対し、本剤を投与する場合には、以下の用法及び用量にて注意して投与すること。[9.3.2 参照],[16.6.2 参照]
7.5.1
軽度の肝機能障害患者(Child-Pugh分類の合計点数:5~6)
- ホスアンプレナビルとして1回700mgを1日2回投与
- ホスアンプレナビルとして1回700mgを1日2回とリトナビル1回100mgを1日1回併用投与
7.5.2
中等度の肝機能障害患者(Child-Pugh分類の合計点数:7~9)
- ホスアンプレナビルとして1回700mgを1日2回投与
7.6
本剤と他の抗HIV薬との併用療法において、因果関係が特定されない重篤な副作用が発現し、治療の継続が困難であると判断された場合には、本剤若しくは併用している他の抗HIV薬の一部を減量又は休薬するのではなく、原則として本剤及び併用している他の抗HIV薬の投与をすべて一旦中止すること。
8. 重要な基本的注意
8.1
本剤はHIV感染症治療の経験を有する医師が投与を行うこと。
* 8.2
本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又は患者に代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。
- 本剤はHIV感染症の根治療法薬ではないことから、日和見感染を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。
8.3
本剤を含む抗HIV薬の多剤併用療法を行った患者で、免疫再構築症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 血友病患者
HIVプロテアーゼ阻害剤で治療中の血友病患者において、皮下血腫、出血性関節症等の出血事象の増加がみられたとの報告がある。[11.1.3参照]
9.1.2 スルホンアミド系薬剤に過敏症の既往歴のある患者
本剤はスルホンアミド基を有するため、交叉過敏症があらわれる可能性がある。
9.3 肝機能障害患者
9.3.1 重度の肝障害患者
投与しないこと。肝臓の代謝機能の低下により、本剤の活性代謝物であるアンプレナビルの高い血中濃度が持続するおそれがある。[2.2 参照],[16.6.2 参照]
9.3.2 肝機能障害のある患者(重度の肝障害患者を除く)
本剤治療前及び治療中は定期的に臨床検査を実施すること。肝炎の患者ではトランスアミナーゼが上昇する危険性がある。肝臓の代謝機能の低下により、高い血中濃度が持続するおそれがある。[7.5 参照],[16.6.2 参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
9.6 授乳婦
授乳を避けさせること。動物実験(ラット)でアンプレナビルは乳汁中へ移行するとの報告がある。また、HIVが乳汁へ移行する可能性がある。
9.7 小児等
低出生体重児、新生児、乳児又は2歳未満の幼児を対象に本剤を投与した臨床試験は実施していない。
9.8 高齢者
患者の肝、腎、及び心機能の低下、合併症、併用薬等を十分考慮し慎重に投与すること。[16.6.4参照]
10. 相互作用
本剤の活性代謝物であるアンプレナビルは、主としてCYP3A4で代謝される。また、アンプレナビルはCYP3A4の阻害作用を有する。[16.4.1 参照],[16.7.1 参照]
10.1 併用禁忌(併用しないこと)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| 治療域が狭くCYP3A4で代謝される薬剤 | アンプレナビルとこれら薬剤はCYP3A4で代謝されるため、併用により代謝が競合的に阻害される。 | |
| シサプリド(国内承認整理済) ピモジド オーラップ [2.3参照] |
これら薬剤の血中濃度が上昇し、不整脈等の重篤な又は生命に危険を及ぼすような事象が起こる可能性がある。 | |
| ベプリジル塩酸塩水和物 ベプリコール [2.3参照] |
ベプリジル塩酸塩水和物の血中濃度が上昇し、生命に危険を及ぼす不整脈が起こる可能性がある。 | |
| ジヒドロエルゴタミンメシル酸塩 ジヒデルゴット等 エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン クリアミン エルゴメトリンマレイン酸塩 メチルエルゴメトリンマレイン酸塩 メテルギン等 [2.3参照] |
これら薬剤の血中濃度が上昇し、末梢血管攣縮、虚血等の重篤な又は生命に危険を及ぼすような事象が起こる可能性がある。 | |
| ミダゾラム ドルミカム等 トリアゾラム ハルシオン等 [2.3参照] |
これら薬剤の血中濃度が上昇し、過度の鎮静や呼吸抑制等の重篤な又は生命に危険を及ぼすような事象が起こる可能性がある。 | |
| バルデナフィル塩酸塩水和物 レビトラ [2.4参照] |
バルデナフィル塩酸塩水和物の血中濃度が上昇し、バルデナフィル塩酸塩水和物に関連する事象(低血圧、失神、視覚障害、持続勃起症等)の発現が増加する可能性がある。 | |
| リファンピシン リファジン、 リマクタン等 [2.5 参照],[16.7.3 参照] |
リファンピシンはアンプレナビルのCmin及びAUCをそれぞれ92%及び82%低下させるため、本剤の作用が減弱する。 | リファンピシンはCYP3A4の強力な誘導剤であるため、アンプレナビルの代謝が促進される。 |
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| エファビレンツ [16.7.2参照] |
本剤1400mg+リトナビル200mg1日1回とエファビレンツ600mg1日1回を併用した場合、アンプレナビルのAUCは13%、Cminは36%低下したが、リトナビルを300mgに増量すると、アンプレナビルの血中濃度は維持された。また、本剤700mg+リトナビル100mg1日2回投与とエファビレンツ600mg1日1回を併用した場合、アンプレナビルの血中濃度に著しい変化はなかった。 | これら薬剤はCYP3A4を誘導するため、本剤の代謝が促進される。 |
| ネビラピン [16.7.2参照] |
本剤1400mg1日2回とネビラピン200mg1日2回を併用した場合、アンプレナビルのCmax、AUC、Cminはそれぞれ25%、33%、35%低下し、ネビラピンのCmax、AUC、Cminはそれぞれ25%、29%、34%上昇した。 また、本剤700mg+リトナビル100mg1日2回とネビラピン200mg1日2回を併用した場合、アンプレナビルのAUC及びCminはそれぞれ11%、19%低下し、ネビラピンのCmax、AUC、Cminはそれぞれ13%、14%、22%上昇した。本剤700mg+リトナビル100mg1日2回とネビラピン200mg1日2回を併用する場合には、用量を調節する必要はない。 なお、本剤+リトナビル1日1回投与におけるネビラピンとの併用試験は実施されていない。 |
|
| インジナビル硫酸塩エタノール付加物 [16.7.3参照] |
アンプレナビルのCmax、AUC、Cminはそれぞれ18%、33%、25%上昇し、インジナビルのCmax、AUC、Cminはそれぞれ22%、38%、27%低下した。 なお、本剤及びリトナビルとインジナビルとの併用における推奨用量は確立していない。 |
アンプレナビルとこれら薬剤はCYP3A4で代謝されるため、併用により代謝が競合的に阻害される。 |
| サキナビル [16.7.3参照] |
アンプレナビルのCmax、AUC、Cminはそれぞれ37%、32%、14%低下し、サキナビルのCmaxは21%上昇し、AUC及びCminはそれぞれ19%、48%低下した。 本剤及びリトナビルとサキナビルとの併用における推奨用量は確立していない。 |
|
| ネルフィナビルメシル酸塩 [16.7.3参照] |
アンプレナビルのCmaxは14%低下し、Cminは189%上昇し、ネルフィナビルのCmax、AUC、Cminはそれぞれ12%、15%、14%上昇した。 本剤及びリトナビルとネルフィナビルとの併用における推奨用量は確立していない。 |
|
| アタザナビル硫酸塩 [16.7.2参照] |
本剤700mg+リトナビル100mg1日2回とアタザナビル300mg1日1回を併用した場合、アタザナビルのCmax、AUCはそれぞれ24%、22%低下した。 なお、本剤及びリトナビルとアタザナビルとの併用における推奨用量は確立していない。 |
|
| ロピナビル・リトナビル [16.7.2参照] |
本剤700mg+リトナビル100mg1日2回とロピナビル・リトナビル(400mg・100mg)1日2回を併用した場合、アンプレナビルのCmax、AUC、Cminはそれぞれ58%、63%、65%低下し、ロピナビルのCmax、AUC、Cminはそれぞれ30%、37%、52%上昇した。 また、本剤1400mg1日2回とロピナビル・リトナビル(533mg・133mg)1日2回を併用した場合、アンプレナビルのCmax、AUC、Cminはそれぞれ13%、26%、42%低下した。 なお、本剤及びリトナビルとロピナビル・リトナビルとの併用における推奨用量は確立していない。 |
|
| リファブチン [16.7.3参照] |
アンプレナビルは、リファブチンのAUCを193%上昇させるため、本剤とリファブチンを併用する場合には、リファブチンの投与量を少なくとも半量に減量し、また、本剤+リトナビルとリファブチンを併用する場合には、リファブチンの投与量を少なくとも1/4に減量し、患者の臨床症状等を十分に観察すること。 | |
| リドカイン(全身投与) アミオダロン塩酸塩 キニジン硫酸塩水和物 シクロスポリンタクロリムス水和物 シロリムス 三環系抗うつ剤 アミトリプチリン、 クロミプラミン等 |
これら薬剤の血中濃度が上昇する可能性があるので、血中濃度のモニタリングを行うことが望ましい。 | |
| ワルファリンカリウム | ワルファリンの血中濃度が上昇する可能性があるので、血液凝固能検査のモニタリングを行うことが望ましい。 | |
| カルシウム拮抗剤 アムロジピン、 ジルチアゼム塩酸塩、 フェロジピン、 ニカルジピン塩酸塩、 ニフェジピン、 ニソルジピン、 ベラパミル塩酸塩等 |
これら薬剤の血中濃度が上昇し、作用が増強する可能性がある。 | |
| ジアゼパム フルラゼパム アルプラゾラム クロラゼプ酸二 カリウム |
これら薬剤の血中濃度が上昇し、作用が増強する可能性がある。 | |
| シンバスタチン アトルバスタチンカルシウム水和物 lovastatin(国内未発売) [16.7.2参照] |
これら薬剤の血中濃度が上昇し、ミオパシー及び横紋筋融解症を起こす可能性がある。本剤700mg+リトナビル100mg1日2回とアトルバスタチン10mg1日1回を併用した場合、アトルバスタチンのCmax、AUC、Cminはそれぞれ184%、153%、73%上昇した。本剤1400mg1日2回とアトルバスタチン10mg1日1回を併用した場合、アンプレナビルのCmax、AUC、Cminはそれぞれ18%、27%、12%低下した。一方、アトルバスタチンのCmax、AUCはそれぞれ304%、130%上昇し、Cminは10%低下した。 本剤と20mg/日以上のアトルバスタチンを併用する場合は、アトルバスタチンの副作用の発現に注意すること。なお、プラバスタチンやフルバスタチンの代謝にはCYP3A4は関与していないため、HMG-CoA還元酵素阻害剤を併用する場合にはプラバスタチンやフルバスタチンが推奨される。 |
|
| シルデナフィル クエン酸塩 タダラフィル |
シルデナフィルクエン酸塩、タダラフィルの血中濃度が上昇し、これら薬剤に関連する事象(低血圧、失神、視覚障害、持続勃起症等)の発現が増加する可能性がある。 よって、併用する場合には、これら薬剤の減量を考慮するとともに、有害事象のモニタリングを行うなど注意すること。 |
|
| ケトコナゾール(経口剤国内未発売) イトラコナゾール [16.7.3参照] |
これら薬剤の血中濃度を上昇させる可能性がある。 特に、高用量のケトコナゾール、イトラコナゾール(>200mg/日)の投与は有益性が危険性を上回ると判断される場合のみ、十分な観察のもとで投与すること。 |
|
| エリスロマイシン クラリスロマイシン |
これら薬剤の血中濃度を上昇させる可能性がある。特に、リトナビルはクラリスロマイシンの血中濃度を上昇させるため、腎障害患者において本剤及びリトナビルとクラリスロマイシンを併用する場合は、クラリスロマイシンの用量を減量すべきである。 | |
| CYP3A4誘導作用を有する抗けいれん薬 フェノバルビタール フェニトイン カルバマゼピン等 |
アンプレナビルの血中濃度を低下させる可能性がある。 | これら薬剤等はCYP3A4を誘導するため、本剤の代謝が促進される。 |
| セイヨウオトギリソウ(St. John’s Wort, セント・ジョーンズ・ワート)含有食品 | アンプレナビルの代謝が促進され血中濃度が低下するおそれがあるので、本剤投与時はセイヨウオトギリソウ含有食品を摂取しないよう注意すること。 | |
| デキサメタゾン | アンプレナビルの血中濃度を低下させる可能性がある。 | |
| ラルテグラビルカリウム [16.7.2参照] |
本剤1400mg1日2回とラルテグラビル400mg1日2回を併用した場合、アンプレナビルのCmax、AUC、Cminはそれぞれ27%、36%、43%低下し、ラルテグラビルのCmax、AUC、Cminはそれぞれ29%、37%、38%低下した(空腹時投与)。 アンプレナビルの血中濃度が低下し、本剤の効果が減弱するおそれがあるので、ブーストしない本剤とラルテグラビルの併用は推奨されない。本剤とラルテグラビルを併用する場合は、さらに低用量のリトナビルも併用すること。 |
機序は不明である。 |
| マラビロク [16.7.2参照] |
本剤700mg+リトナビル100mg1日2回とマラビロク300mg1日2回を併用した場合、アンプレナビルのCminは36%減少し、マラビロクのAUCは149%増加した。また、本剤1400mg+リトナビル100mg1日1回とマラビロク300mg1日1回を併用した場合、アンプレナビルのCminは15%減少し、マラビロクのAUCは126%増加した。 本剤+リトナビルとマラビロクを併用する場合には、マラビロクの用量を150mg1日2回に減量すること。 |
本剤及びリトナビルのCYP3A4に対する阻害作用により、マラビロクの代謝が阻害される。 |
| イブルチニブ | イブルチニブの血中濃度が上昇する可能性がある。 | 本剤のCYP3A4に対する阻害作用により、イブルチニブの代謝が阻害される。 |
| エベロリムス | エベロリムスの血中濃度が上昇する可能性がある。 | 本剤のCYP3A4に対する阻害作用により、エベロリムスの代謝が阻害される。 |
| テラプレビル | 本剤+リトナビルとテラプレビルの併用により、定常状態におけるアンプレナビルとテラプレビルの血中濃度が低下する可能性がある。 | 機序は不明である。 |
| シメプレビル | シメプレビルの血中濃度が変化する可能性がある。シメプレビルの血中濃度が低下しシメプレビルの効果が減弱する、もしくはシメプレビルの血中濃度が上昇し副作用が発現するおそれがあるので、本剤とシメプレビルを併用する場合は十分注意すること。 | 本剤のCYP3A4阻害作用又は誘導作用により、シメプレビルの代謝が阻害又は促進される。 |
| ドルテグラビルナトリウム [16.7.2参照] |
本剤700mg+リトナビル100mg1日2回とドルテグラビル50mg1日1回の併用により、ドルテグラビルの血漿中濃度がCmaxで24%、Cτで49%低下させたとの報告があるため、HIVインテグラーゼ阻害剤に耐性を有する患者では、本剤と併用しないこと。 | 本剤がCYP3A4及びUGT1A1を誘導することにより、ドルテグラビルの代謝が促進される。 |
| パロキセチン塩酸塩水和物 | パロキセチン塩酸塩水和物の作用が減弱する可能性がある。本剤及びリトナビルとパロキセチン塩酸塩水和物の併用により、パロキセチンの血中濃度が約60%低下したとの報告がある。 | 機序は不明である。 |
| 黄体・卵胞ホルモン剤 エチニルエストラジオール、ノルエチステロン等 [16.7.2参照] |
本剤及びリトナビルと黄体・卵胞ホルモン剤の併用により、リトナビルの血中濃度の上昇及び黄体・卵胞ホルモン剤の血中濃度の低下がみられ、肝トランスアミナーゼの上昇や黄体・卵胞ホルモンレベルの変動がみられる可能性がある。 また、本剤投与時に避妊する場合は、これらの経口避妊薬とは別の避妊法を行うことが望ましい。なお、本剤及びリトナビルと高用量のエストロゲンやプロゲストゲンを併用した場合のデータは得られておらず、有効性・安全性は確立していない。 |
機序は不明である。 |
11. 副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1 重大な副作用
11.1.1 皮膚粘膜眼症候群(Stevens-Johnson症候群)(1%未満)
Stevens-Johnson症候群等の重度又は生命に危険を及ぼすような発疹があらわれたとの報告がある。重度の発疹、及び全身的症状又は粘膜症状を伴う中等度の発疹があらわれた場合は、本剤の投与を直ちに中止し適切な処置を行うこと。
11.1.2 高血糖(1%未満)、糖尿病(頻度不明)
糖尿病・糖尿病の悪化、糖尿病性ケトアシドーシス及び高血糖があらわれることがあるので、このような症状があらわれた場合は、インスリン又は経口糖尿病薬の投与開始や用量調節など適切な処置を行うこと。
11.1.3 出血傾向(頻度不明)
皮下血腫、出血性関節症等の出血事象の増加があらわれることがあるので、本剤投与中は出血事象の増加に注意し、このような症状があらわれた場合は、血液凝固因子を投与するなど適切な処置を行うこと。[9.1.1参照]
11.1.4 横紋筋融解症、筋炎、CK上昇(いずれも頻度不明)、筋痛(1%未満)
特にHIV逆転写酵素阻害剤を併用している患者においてあらわれたとの報告がある。
11.2 その他の副作用
| 1%~10%未満 | 1%未満 | 頻度不明 | |
|---|---|---|---|
| 皮膚 | 発疹、そう痒 | 紅斑、斑状丘疹性皮疹 | 血管浮腫 |
| 精神神経系 | 頭痛 | ||
| 心臓障害 | 心筋梗塞 | ||
| 消化器 | 下痢、悪心、嘔吐、腹痛 | 鼓腸、口の錯感覚 | |
| 肝臓 | 肝機能検査値異常(AST、ALT等の上昇) | ||
| 代謝・栄養障害 | 高脂血症体脂肪の再分布/蓄積(胸部、体幹部の脂肪増加、末梢部、顔面の脂肪減少、野牛肩、血清脂質増加、血糖増加)、リパーゼ上昇 | インスリン抵抗性 | |
| 腎及び尿路障害 | 腎結石症 | ||
| 全身症状 | 疲労 |
13. 過量投与
13.1 処置
アンプレナビルは高い蛋白結合率を有するため、腹膜透析や血液透析により除去できる可能性は低い。[16.3.2参照]
15. その他の注意
15.2 非臨床試験に基づく情報
15.2.1
長期がん原性試験(104週)において、雄マウス(250mg/kg/日以上)で肝細胞腺腫及び肝細胞癌並びに雌雄ラット(各々、825及び300mg/kg/日以上)で肝細胞腺腫及び甲状腺濾胞細胞腺腫の増加がみられた。
なお、ラットの反復投与試験において、甲状腺濾胞細胞腺腫の発現に関与する肝薬物代謝酵素誘導を示唆する所見がみられた。また、ラットでは対照群に比べ精巣間細胞過形成(825mg/kg/日以上)及び子宮内膜腺癌(2250mg/kg/日)の軽度な増加がみられたが、子宮内膜腺癌の発現率は背景値範囲内であった。臨床試験や市販後の使用経験からは、これら所見が臨床的に重要であることを示唆する報告は得られていない。
なお、マウス(250~600mg/kg/日)及びラット(300~2250mg/kg/日)のがん原性試験における曝露量は、ヒトに本剤1400mg1日2回投与した場合の曝露量の0.3~0.7倍及び0.7~1.4倍、本剤1400mg及びリトナビル200mg1日1回投与した場合の曝露量の0.2~0.3倍及び0.3~0.7倍、本剤700mg及びリトナビル100mg1日2回投与した場合の曝露量の0.1~0.3倍及び0.3~0.6倍に相当する。
15.2.2
イヌの反復投与試験において、流涎、嘔吐、軟便あるいは液状便がみられ、脱水及び電解質喪失が観察された。また、ラット及びイヌにおいて、肝酵素の上昇、肝重量の増加、肝細胞壊死等が報告されている。
15.2.3
In vitro及びin vivo試験である細菌を用いる復帰突然変異試験、マウスリンフォーマTK試験、ラット小核試験あるいはヒトリンパ球を用いる染色体異常試験において、本薬は遺伝毒性を示さなかった。
16. 薬物動態
16.1 血中濃度
16.1.1 単回経口投与
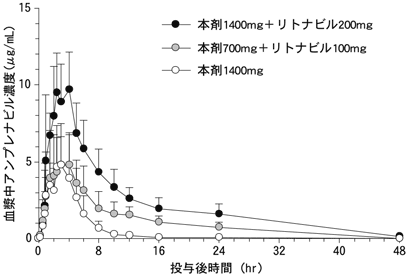
健康成人男性9例に本剤1400mg、本剤1400mgとリトナビル200mg、あるいは本剤700mgとリトナビル100mgを単回経口投与した時の血漿中アンプレナビル濃度推移を図-1に、アンプレナビルの薬物動態パラメータを表-1に示した。アンプレナビルは投与約3時間後に最高血漿中濃度に達し、消失半減期は約6~7時間であった。
| 投与方法 | Cmax (μg/mL) |
Tmax (h) |
t1/2 (h) |
AUC0-∞ (μg・h/mL) |
|---|---|---|---|---|
| 本剤1400mg | 5.79±2.41 | 3.08±1.35 | 6.47±2.98 | 23.05±7.83 |
| 本剤1400mg+ リトナビル200mg |
11.12±2.64 | 3.17±1.12 | 7.19±1.86 | 101.20±27.94 |
| 本剤700mg+ リトナビル100mg |
6.69±1.89 | 2.89±1.54 | 6.16±1.16 | 50.65±14.49 |
平均値±標準偏差、9例
16.1.2 反復経口投与
健康成人に本剤1400mgを1日2回、本剤1400mgとリトナビル200mgを1日1回、あるいは本剤700mgとリトナビル100mgを1日2回反復経口投与した時の定常状態におけるアンプレナビルの薬物動態パラメータは下記の通りであった(外国人データ)。
| 投与方法 | n | Cmax (μg/mL) |
Tmax (h) |
AUC0-24 (μg・h/mL) |
Cmin (μg/mL) |
|---|---|---|---|---|---|
| 本剤1400mg1日2回 | 12 | 4.91±1.94 | 1.8±0.7 | 36.4±12.6注1) | 0.25±0.12 |
| 本剤1400mg1日1回 +リトナビル 200mg1日1回 |
70 | 7.56±2.01 | 2.3±1.1 | 67.3±18.2 | 1.33±0.57 |
| 本剤700mg1日2回 +リトナビル 100mg1日2回 |
24 | 6.31±1.72 | 2.0±1.3 | 83.2±27.4注1) | 2.33±1.13 |
平均値±標準偏差
注1)AUC0-12を2倍した値
健康成人に本剤1400mgとリトナビル100mgあるいは200mgを、同一被験者にクロスオーバー法により1日1回、14日間併用投与した時の定常状態におけるアンプレナビルの薬物動態パラメータは以下の通りであった。本剤1400mgに併用するリトナビルの用量にかかわらず、アンプレナビルの薬物動態は同様であった(外国人データ)。
| 投与方法 | n | Cmax (μg/mL) |
Tmax注1) (h) |
AUC0-24 (μg・h/mL) |
Cmin (μg/mL) |
|---|---|---|---|---|---|
| 本剤1400mg+ リトナビル100mg |
36 | 7.93 (7.25-8.68) |
1.50 (0.75-5.00) |
66.36 (61.06-72.12) |
0.86 (0.74-1.01) |
| 本剤1400mg+ リトナビル200mg |
36 | 8.17 (7.58-8.81) |
2.00 (1.00-4.00) |
73.80 (66.93-81.37) |
1.40 (1.19-1.65) |
幾何平均値(95%信頼区間)
注1)中央値(範囲)
16.1.3 健康成人とHIV感染症患者の比較
健康成人とHIV感染症患者を対象とした各薬物動態試験において、本剤及びリトナビル投与後のアンプレナビルの薬物動態に差は認められなかった(外国人データ)。
16.2 吸収
16.2.1 食事の影響
健康成人に本剤1400mgを経口投与した時、食事による薬物動態への影響はなかった(外国人データ)。
16.3 分布
16.3.1 分布容積
健康成人に本剤を経口投与した時のアンプレナビルの見かけの分布容積は約600Lであった。リトナビルとの併用投与により、アンプレナビルの見かけの分布容積は約40%低下した(外国人データ)。
16.3.2 蛋白結合率
In vitroで、アンプレナビルの血漿蛋白結合率は約90%であった。[13.1参照]
16.4 代謝
16.4.1 主な代謝酵素
本剤は経口投与後、主に消化管上皮において速やかにアンプレナビルと無機リン酸に加水分解される。アンプレナビルは、主に肝臓においてCYP3A4により代謝される。[10.参照]
16.4.2 In vivo試験
本剤とリトナビルを併用投与した場合、リトナビルによる強力なCYP3A4阻害により、アンプレナビルの代謝が阻害される結果、血漿中アンプレナビル濃度が上昇する(外国人データ)。なお、本剤との併用投与で使用されるリトナビルの用量(100mg1日2回あるいは200mg1日1回投与)は、リトナビルの通常臨床用量(600mg1日2回投与)の6分の1であり、本剤とリトナビルを併用投与した場合の抗ウイルス活性は主にアンプレナビルによるものである。[18.1参照]
16.5 排泄
HIV感染症患者に本剤を経口投与した時のアンプレナビルの消失半減期は約8.5時間であった。本剤とリトナビルを併用投与した時、アンプレナビルの消失半減期は12時間に延長した。アンプレナビルの主要な消失経路は肝代謝であり、未変化体(アンプレナビル)の尿中排泄率は1%未満であった。健康成人に14C-アンプレナビルを単回経口投与した時の代謝物及び未変化体の尿中排泄率は約14%、糞中排泄率は約75%であった(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
腎機能障害患者における検討は行われていないが、腎排泄はアンプレナビルあるいはリトナビルの主要排泄経路ではなく、これらの排泄に対する腎機能障害の影響は少ないと考えられるため、用量調節を行う必要はない。
16.6.2 肝機能障害患者
(1)
軽度の肝機能障害(Child-Pugh分類の合計点数:5~6)を有するHIV感染症患者に本剤700mg1日2回+リトナビル100mg1日1回を併用投与した結果、肝機能の正常なHIV感染症患者に本剤700mg1日2回+リトナビル100mg1日2回を併用投与した場合と比較して、血漿中アンプレナビルのCmax及びAUC0-∞はわずかに高く(それぞれ17%及び22%高い)、Cτは同様であった(外国人データ)。
(2)
中等度の肝機能障害(Child-Pugh分類の合計点数:7~9)を有するHIV感染症患者に本剤700mg1日1回+リトナビル100mg1日1回を併用投与した結果、肝機能の正常なHIV感染症患者に本剤700mg1日2回+リトナビル100mg1日2回を併用投与した場合と比較して、血漿中アンプレナビルのCavg、Cτ及び非結合アンプレナビルのCτは、それぞれ24%、65%及び42%低かった(外国人データ)。
(3)
重度の肝機能障害(Child-Pugh分類の合計点数:10~13)を有するHIV感染症患者に本剤300mg注)1日2回+リトナビル100mg1日1回を併用投与した結果を本剤700mgに標準化し、肝機能の正常なHIV感染症患者に本剤700mg1日2回+リトナビル100mg1日2回を併用投与した場合と比較すると、アンプレナビルのAUC0-τが約80%増加することが示された(外国人データ)。
(4)
肝機能障害患者に本剤を単独投与した時の薬物動態成績は得られていない。しかしながら、肝機能障害患者に対するアンプレナビル600mgの単回経口投与において、中等度の肝硬変患者のAUC0-∞(25.76μg・h/mL)は、健康成人(12.00μg・h/mL)と比較して有意に高値を示した。また、重度の肝硬変患者のAUC0-∞及びCmax(AUC0-∞:38.66μg・h/mL、Cmax:9.43μg/mL)は、健康成人(AUC0-∞:12.00μg・h/mL、Cmax:4.90μg/mL)と比較して有意に高値を示した(外国人データ)。[2.2 参照],[7.5 参照],[9.3.1 参照],[9.3.2 参照]
16.6.3 小児等
2~18歳のHIV感染症患者におけるアンプレナビルの薬物動態は成人と同様であった(外国人データ)。
これらの患者における定常状態時のアンプレナビルの薬物動態パラメータを以下に示す。
| 2-5歳 | 6-11歳 | 12-18歳 | ||||
|---|---|---|---|---|---|---|
| n | FPV注1) 30mg/kg注) |
n | FPV注1) 18mg/kg注) +RTV注2) 3mg/kg |
n | FPV注1) 700mg注) +RTV注2) 100mg |
|
| AUC0-24 (μg・h/mL) |
8 | 31.4 (13.7-72.4) |
9 | 93.4 (67.8-129) |
8 | 58.8 (38.8-89.0) |
| Cmax (μg/mL) |
8 | 5.00 (1.95-12.8) |
9 | 6.07 (4.40-8.38) |
8 | 4.33 (2.82-6.65) |
| Cτ (μg/mL) |
17 | 0.454 (0.342-0.604) |
17 | 2.69 (2.15-3.36) |
24 | 1.61 (1.21-2.15) |
幾何平均(95%信頼区間)
注1)FPV:ホスアンプレナビル
注2)RTV:リトナビル
16.6.4 高齢者
65歳以上の患者において、本剤投与時の薬物動態の検討は行われていない。[9.8参照]
16.7 薬物相互作用
16.7.1 In vitro試験
アンプレナビルはCYP3A4の阻害作用を有する。[10.参照]
16.7.2 本剤と各種薬剤の併用による薬物動態への影響
本剤又は本剤とリトナビルを併用薬と投与した時の薬物動態パラメータの変化を以下に示す(外国人データ)。
| 併用薬 | 併用薬の用量 (mg) |
本剤の用量 (mg) |
n | アンプレナビルの薬物動態 パラメータの変化率(%) |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| 抗ウイルス化学療法剤(非ヌクレオシド系逆転写酵素阻害剤) | ||||||
| エファビレンツ [10.2参照] |
600 QD, 2週間 |
1400 QD +リトナビル200 QD, 2週間 |
16 | ⇔ | ↓13 | ↓36 |
| エファビレンツ [10.2参照] |
600 QD, 2週間 |
700 BID +リトナビル100 BID, 2週間 |
16 | ⇔ | ⇔ | ↓17 |
| ネビラピン [10.2参照] |
200 BID, 2週間注1) | 1400 BID, 2週間 |
17 | ↓25 | ↓33 | ↓35 |
| ネビラピン [10.2参照] |
200 BID, 2週間注1) |
700 BID +リトナビル100 BID, 2週間 |
17 | ⇔ | ↓11 | ↓19 |
| 抗ウイルス化学療法剤(HIVプロテアーゼ阻害剤) | ||||||
| アタザナビル | 300 QD,10日間 | 700 BID +リトナビル100 BID, 10日間 |
22 | ⇔ | ⇔ | ⇔ |
| ロピナビル・ リトナビル [10.2参照] |
533・133 BID, 2週間 |
1400 BID, 2週間 |
18 | ↓13注2) | ↓26注2) | ↓42注2) |
| ロピナビル・ リトナビル [10.2参照] |
400・100 BID, 2週間 |
700 BID +リトナビル100 BID, 2週間 |
18 | ↓58 | ↓63 | ↓65 |
| 抗ウイルス化学療法剤(HIVインテグラーゼ阻害剤) | ||||||
| ラルテグラビル (空腹時) [10.2参照] |
400 BID, 2週間 |
1400 BID, 2週間 |
14 | ↓27 | ↓36 | ↓43 |
| 抗ウイルス化学療法剤(CCR5阻害剤) | ||||||
| マラビロク [10.2参照] |
300 BID, 10日間 |
700 BID +リトナビル100 BID, 10日間 |
14 | ↓34 | ↓35 | ↓36 |
| マラビロク [10.2参照] |
300 QD, 10日間 |
1400 QD +リトナビル100 QD, 10日間 |
14 | ↓29 | ↓30 | ↓15 |
| 制酸剤 | ||||||
| 水酸化アルミニウム・ 水酸化マグネシウム |
3600・1800 (30mL), 単回 |
1400,単回 | 30 | ↓35 | ↓18 | ↑14 |
| H2受容体拮抗剤 | ||||||
| ラニチジン | 300,単回 | 1400,単回 | 30 | ↓51 | ↓30 | ⇔ |
| HMG-CoA還元酵素阻害剤 | ||||||
| アトルバスタチン | 10 QD,4日間 | 700 BID +リトナビル100 BID, 2週間 |
16 | ⇔ | ⇔ | ⇔ |
| アトルバスタチン [10.2参照] |
10 QD,4日間 | 1400 BID, 2週間 |
16 | ↓18 | ↓27 | ↓12 |
| 抗けいれん薬 | ||||||
| フェニトイン | 300 QD,10日間 | 700 BID +リトナビル100 BID, 10日間 |
13 | ⇔ | ↑20 | ↑19 |
| 経口避妊薬 | ||||||
| エチニルエストラジオール・ ノルエチステロン注3) |
0.035・0.5 QD, 21日間 |
700 BID +リトナビル注4) 100 BID,21日間 |
25 | ⇔ | ⇔ | ⇔ |
↑:上昇,↓:低下,⇔:変化なし(変化率が10%未満)
QD:1日1回投与,BID:1日2回投与
注1)試験開始前に少なくとも12週間はネビラピンを投与していた。
注2)本剤700mg BID+リトナビル100mg BID投与時との比較
注3)既存データとの比較
注4)リトナビルのCmax、AUC、Cminは、それぞれ63%、45%、13%上昇した(他の臨床試験で得られたデータとの比較)。
| 併用薬 | 併用薬の用量 (mg) |
本剤の用量 (mg) |
n | 併用薬の薬物動態パラメータの変化率(%) | ||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| 抗ウイルス化学療法剤(非ヌクレオシド系逆転写酵素阻害剤) | ||||||
| ネビラピン [10.2参照] |
200 BID, 2週間注1) | 1400 BID, 2週間 |
17 | ↑25 | ↑29 | ↑34 |
| ネビラピン [10.2参照] |
200 BID, 2週間注1) |
700 BID +リトナビル100 BID, 2週間 |
17 | ↑13 | ↑14 | ↑22 |
| 抗ウイルス化学療法剤(HIVプロテアーゼ阻害剤) | ||||||
| アタザナビル [10.2 参照] |
300 QD,10日間 | 700 BID +リトナビル100 BID, 10日間 |
21 | ↓24 | ↓22 | ⇔ |
| ロピナビル・ リトナビル [10.2参照] |
533・133 BID, 2週間 |
1400 BID, 2週間 |
18 | ⇔注2),注3) | ⇔注2),注3) | ⇔注2),注3) |
| ロピナビル・ リトナビル [10.2参照] |
400・100 BID, 2週間 |
700 BID +リトナビル100 BID, 2週間 |
18 | ↑30注2) | ↑37注2) | ↑52注2) |
| 抗ウイルス化学療法剤(HIVインテグラーゼ阻害剤) | ||||||
| ラルテグラビル (空腹時) [10.2参照] |
400 BID, 2週間 |
1400 BID, 2週間 |
14 | ↓29 | ↓37 | ↓38 |
| ドルテグラビル4) [10.2 参照] |
50 QD, 15日間 |
700 BID +リトナビル100 BID, 10日間 |
14 | ↓29 | ↓37 | ↓38 |
| 抗ウイルス化学療法剤(CCR5阻害剤) | ||||||
| マラビロク [10.2参照] |
300 BID, 10日間 |
700 BID +リトナビル100 BID, 10日間 |
14 | ↑52 | ↑149 | ↑374 |
| マラビロク [10.2参照] |
300 QD, 10日間 |
1400 QD +リトナビル100 QD, 10日間 |
14 | ↑45 | ↑126 | ↑80 |
| HMG-CoA還元酵素阻害剤 | ||||||
| アトルバスタチン[10.2 参照] | 10 QD,4日間 | 700 BID +リトナビル100 BID, 2週間 |
16 | ↑184 | ↑153 | ↑73 |
| アトルバスタチン [10.2参照] |
10 QD,4日間 | 1400 BID, 2週間 |
16 | ↑304 | ↑130 | ↓10 |
| 抗けいれん薬 | ||||||
| フェニトイン | 300 QD,10日間 | 700 BID +リトナビル100 BID, 10日間 |
14 | ↓20 | ↓22 | ↓29 |
| オピオイド受容体作動薬 | ||||||
| メサドン | 200以下 QD,14日間 | 700 BID+リトナビル 100 BID,14日間 |
19 | R-methadone(活性体) | ||
| ↓21 | ↓18 | ↓11 | ||||
| 非結合R-methadone 2時間後血中濃度:⇔ 6時間後血中濃度:⇔ |
||||||
| S-methadone(不活性体) | ||||||
| ↓43 | ↓43 | ↓41 | ||||
| 非結合R-methadone 2時間後血中濃度:⇔ 6時間後血中濃度:↓19 |
||||||
| 経口避妊薬 | ||||||
| エチニルエストラジオール(EE) ・ノルエチステロン(NE) [10.2 参照] |
0.035・0.5 QD, 21日間 |
700 BID +リトナビル100 100 BID,21日間 |
25 | EE:↓28 NE:↓38 |
EE:↓37 NE:↓34 |
EE:ND NE:↓26 |
↑:上昇,↓:低下,⇔:変化なし(変化率が10%未満)
ND:定量限界以下であったため評価せず
QD:1日1回投与,BID:1日2回投与
注1)試験開始前に少なくとも12週間はネビラピンを投与していた。
注2)ロピナビルの薬物動態パラメータの変化率
注3)ロピナビル400mg・リトナビル100mg BID投与時との比較
16.7.3 アンプレナビルと各種薬剤の併用による薬物動態への影響
本剤は体内においてアンプレナビルに変換されるため、参考としてアンプレナビルを各種薬剤と併用投与した時の薬物動態パラメータの変化を以下に示す。
| 併用薬 | 併用薬の用量 (mg) |
アンプレナビルの用量 (mg) |
n | アンプレナビルの薬物動態 パラメータの変化率(%) |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| 抗ウイルス化学療法剤(ヌクレオシド系逆転写酵素阻害剤) | ||||||
| ラミブジン | 150,単回 | 600,単回 | 11 | ⇔ | ⇔ | NA |
| ジドブジン | 300,単回 | 600,単回 | 12 | ⇔ | ↑13 | NA |
| 抗ウイルス化学療法剤(非ヌクレオシド系逆転写酵素阻害剤) | ||||||
| デラビルジン | 600 BID,10日間 | 600 BID,10日間 | 9 | ↑40 | ↑130 | ↑125 |
| 抗ウイルス化学療法剤(HIVプロテアーゼ阻害剤) | ||||||
| インジナビル [10.2参照] |
800 TID, 2週間(空腹時) |
750又は800 TID, 2週間(空腹時) |
9 | ↑18 | ↑33 | ↑25 |
| サキナビル [10.2参照] |
800 TID, 2週間(食後) |
750又は800 TID, 2週間(食後) |
7 | ↓37 | ↓32 | ↓14 |
| ネルフィナビル [10.2参照] |
750 TID, 2週間(食後) |
750又は800 TID, 2週間(食後) |
6 | ↓14 | ⇔ | ↑189 |
| 感染症治療剤 | ||||||
| クラリスロマイシン | 500 BID, 4日間 |
1200 BID,4日間 | 12 | ↑15 | ↑18 | ↑39 |
| ケトコナゾール | 400,単回 | 1200,単回 | 12 | ↓16 | ↑31 | NA |
| リファンピシン [2.5 参照],[10.1 参照] |
300 QD,4日間 | 1200 BID,4日間 | 11 | ↓70 | ↓82 | ↓92 |
| リファブチン | 300 QD,10日間 | 1200 BID,10日間 | 6 | ⇔ | ↓15 | ↓15 |
↑:上昇, ↓:低下, ⇔:変化なし(変化率が10%未満)
NA:単回投与試験のため算出せず
BID:1日2回投与, TID:1日3回投与
| 併用薬 | 併用薬の用量 (mg) |
アンプレナビルの用量 (mg) |
n | 併用薬の薬物動態 パラメータの変化率(%) |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| 抗ウイルス化学療法剤(ヌクレオシド系逆転写酵素阻害剤) | ||||||
| ラミブジン | 150,単回 | 600,単回 | 11 | ⇔ | ⇔ | NA |
| ジドブジン | 300,単回 | 600,単回 | 12 | ↑40 | ↑31 | NA |
| 抗ウイルス化学療法剤(非ヌクレオシド系逆転写酵素阻害剤) | ||||||
| デラビルジン | 600 BID, 10日間 | 600 BID, 10日間 | 9 | ↓47 | ↓61 | ↓88 |
| 抗ウイルス化学療法剤(HIVプロテアーゼ阻害剤) | ||||||
| インジナビル注1)[10.2参照] | 800 TID, 2週間 (空腹時) |
750又は800 TID, 2週間 (空腹時) |
9 | ↓22 | ↓38 | ↓27 |
| サキナビル注1) [10.2参照] |
800 TID, 2週間 (食後) |
750又は800 TID, 2週間 (食後) |
7 | ↑21 | ↓19 | ↓48 |
| ネルフィナビル注1) [10.2参照] |
750 TID, 2週間 (食後) |
750又は800 TID, 2週間 (食後) |
6 | ↑12 | ↑15 | ↑14 |
| 感染症治療剤 | ||||||
| クラリスロマイシン | 500 BID, 4日間 | 1200 BID, 4日間 | 12 | ↓10 | ⇔ | ⇔ |
| ケトコナゾール [10.2参照] |
400, 単回 | 1200, 単回 | 12 | ↑19 | ↑44 | NA |
| リファンピシン | 300 QD,4日間 | 1200 BID,4日間 | 11 | ⇔ | ⇔ | ND |
| リファブチン [10.2参照] |
300 QD,10日間 | 1200 BID,10日間 | 6 | ↑119 | ↑193 | ↑271 |
↑:上昇, ↓:低下, ⇔:変化なし(変化率が10%未満)
NA:単回投与試験のため算出せず
ND:定量限界以下であったため評価せず
BID:1日2回投与, TID:1日3回投与
注1)既存データとの比較
注)本剤の承認された用法及び用量は、「通常、成人には、抗HIV薬の治療経験がない患者の場合、ホスアンプレナビルとして1回700mgとリトナビル1回100mgをそれぞれ1日2回併用投与、ホスアンプレナビルとして1回1400mgとリトナビル1回100mg又は200mgをそれぞれ1日1回併用投与、又はホスアンプレナビルとして1回1400mgを1日2回投与、HIVプロテアーゼ阻害剤の投与経験がある患者の場合、ホスアンプレナビルとして1回700mgとリトナビル1回100mgをそれぞれ1日2回併用投与」である。投与に際しては、必ず他の抗HIV薬と併用すること。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第III相臨床試験(APV30002)
抗HIV薬の治療経験がない18歳以上の患者649例を対象とした無作為オープン比較試験(アバカビル300mg1日2回とラミブジン150mg1日2回の併用による、本剤1400mg1日1回+リトナビル200mg1日1回投与群322例又はネルフィナビル1250mg1日2回投与群327例)において、48週間の治療期間中に血漿中のHIV-1 RNA量が検出限界(400copies/mL)未満であった患者の推移を図-1に示した。48週間の治療により、検出限界未満の患者の比率は本剤+リトナビル投与群で69%、ネルフィナビル投与群で68%となり、同等であった。血漿中HIV-1 RNA量が50copies/mL未満の患者の比率もそれぞれ58%、55%であった。48週間の治療後のCD4リンパ球数の増加量(中央値)は、本剤+リトナビル群で203/mm3、ネルフィナビル群で207/mm3であった。
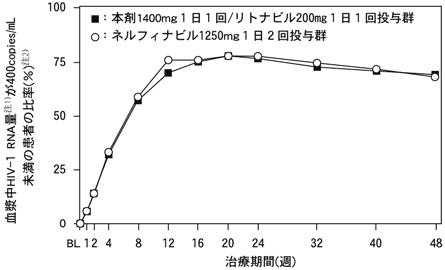
注1)Roche AMPLICOR MONITOR assay(Version 1.5)
注2)中断、データ欠測、リバウンドはHIV-1 RNA量が400copies/mL以上とみなした
なお、本試験における試験成績の要約を表-1に示した。
| 結果 | 本剤1400mg リトナビル200mg 1日1回 + アバカビル+ (n=322) |
ネルフィナビル1250mg + アバカビル+ (n=327) |
|---|---|---|
| レスポンダー注1) | 69% (58%) |
68% (55%) |
| ウイルス学的な治療失敗注2) | 6% | 16% |
| 死亡 | 1% | 0% |
| 有害事象による中止 | 9% | 6% |
| その他の理由による中止注3) | 15% | 10% |
(n=Intent-to-treat-analysis)
注1)血漿中HIV-1 RNA量が400copies/mL未満、( )内の値は血漿中HIV-1 RNA量が50copies/mL未満
注2)リバウンドを起こした患者、48週を通じてHIV-1 RNA量が減少しなかった患者
注3)同意の撤回、試験途中でフォローアップ不可、プロトコール違反、データ欠測等
本剤1400mg1日1回及びリトナビル200mg1日1回を48週間投与した際にみられた副作用発現頻度は73%(234/322例)であった。主な副作用は、悪心31%(100/322例)、下痢29%(93/322例)、嘔吐15%(47/322例)、疲労10%(32/322例)及び頭痛10%(31/322例)であった(外国人データ)。
17.1.2 海外第III相臨床試験(APV30003)
ウイルス学的な治療失敗を伴うHIVプロテアーゼ阻害剤の投与経験(2種類以下)がある患者315例を対象とした無作為オープン比較試験において、本剤700mg1日2回+リトナビル100mg1日2回投与群107例、本剤1400mg1日1回+リトナビル200mg1日1回投与群105例、ロピナビル400mg+リトナビル100mg1日2回投与群103例を比較した。前治療としての逆転写酵素阻害剤の治療期間の中央値(3種類以上の治療経験患者の割合)はそれぞれ257週間(79%)、234週間(70%)、210週間(64%)であり、HIVプロテアーゼ阻害剤の治療期間の中央値(2種類以上の治療経験患者の割合)はそれぞれ149週間(49%)、149週間(57%)、130週間(40%)であった。48週間の治療により、血漿中のHIV-1 RNA量が400copies/mL未満(50copies/mL未満)であった患者の比率は本剤1日2回投与群で58%(46%)、本剤1日1回投与群で50%(37%)、ロピナビル投与群で61%(50%)であった。また、48週間の治療後のCD4リンパ球数の増加量(中央値)はそれぞれ81/mm3、61/mm3、91/mm3あった。
なお、本試験における試験成績の要約を表-2に示した。
| 結果 | 本剤700mg + リトナビル100mg (n=107) |
本剤1400mg + リトナビル200mg (n=105) |
ロピナビル400mg + リトナビル100mg (n=103) |
|---|---|---|---|
| レスポンダー注1) | 58% (46%) |
50% (37%) |
61% (50%) |
| ウイルス学的な治療失敗注2) | 29% | 41% | 27% |
| 死亡 | <1% | 0% | 0% |
| 有害事象による中止 | 5% | 2% | 7% |
| その他の理由による中止注3) | 9% | 8% | 5% |
(n=Intent-to-treat-analysis)
注1)血漿中HIV-1 RNA量が400copies/mL未満、( )内の値は血漿中HIV-1 RNA量が50copies/mL未満
注2)リバウンドを起こした患者、ウイルス学的効果が不十分のため投与を中止した患者、48週を通じてHIV-1 RNA量が減少しなかった患者
注3)同意の撤回、試験途中でフォローアップ不可、プロトコール違反等
本剤700mg1日2回及びリトナビル100mg1日2回を投与した際にみられた副作用発現頻度は55%(58/106例)で、主な副作用は、下痢23%(24/106例)、悪心15%(16/106例)及び頭痛10%(11/106例)であった。本剤1400mg1日1回及びリトナビル200mg1日1回を投与した際にみられた副作用発現頻度は51%(54/106例)で、主な副作用は、下痢23%(24/106例)、悪心17%(18/106例)及び嘔吐10%(11/106例)であった(外国人データ)。
17.1.3 海外第III相臨床試験(APV30001)
抗HIV薬の治療経験がない17歳以上の患者249例を対象とした無作為オープン比較試験(アバカビル300mg1日2回とラミブジン150mg1日2回の併用による、本剤1400mg1日2回投与群166例又はネルフィナビル1250mg1日2回投与群83例)において、48週間の治療により、血漿中HIV-1 RNA量が検出限界未満(400copies/mL未満)の患者の比率は本剤投与群で66%、ネルフィナビル投与群で52%であった。血漿中HIV-1 RNA量が50copies/mL未満の患者の比率ではそれぞれ57%、42%であった。48週間の治療後のCD4リンパ球数の増加量(中央値)は、本剤群で201/mm3、ネルフィナビル群で216/mm3であった。
なお、本試験における試験成績の要約を表-3に示した。
| 結果 | 本剤1400mg + アバカビル+ (n=166) |
ネルフィナビル1250mg + アバカビル+ (n=83) |
|---|---|---|
| レスポンダー注1) | 66% (57%) |
52% (42%) |
| ウイルス学的な治療失敗注2) | 19% | 32% |
| 症状の進行 | 1% | 1% |
| 死亡 | 0% | 1% |
| 有害事象による中止 | 4% | 2% |
| その他の理由による中止注3) | 10% | 10% |
(n=Intent-to-treat-analysis)
注1)血漿中HIV-1 RNA量が400copies/mL未満、( )内の値は血漿中HIV-1 RNA量が50copies/mL未満
注2)リバウンドを起こした患者、48週を通じて血漿中HIV-1 RNA量が減少しなかった患者
注3)同意の撤回、試験途中でフォローアップ不可、プロトコール違反、データ欠測等
本剤(1400mg1日2回)を、アバカビル(300mg1日2回)及びラミブジン(150mg1日2回)と48週間併用投与した際にみられた副作用発現頻度は61%(101/166例)であった。主な副作用は、悪心31%(51/166例)、下痢17%(29/166例)、嘔吐12%(20/166例)、発疹12%(20/166例)及び薬物過敏症10%(16/166例)であった(外国人データ)。
17.1.4 海外第II相臨床試験(APV20003、APV29005)(2~18歳)
2~18歳のHIV感染症患者144例を対象とした2つのオープン試験において、これらの患者における本剤の有効性及び安全性が確認された。本剤及びリトナビルの用量については、患者の体重及び年齢に基づき設定した。
上記2試験において、本剤のみを投与された症例はHIVプロテアーゼ阻害剤の投与経験がない患者18例(うち16例は抗HIV薬の治療経験がない患者)、本剤+リトナビルが併用投与された症例はHIVプロテアーゼ阻害剤の投与経験がない患者59例及びHIVプロテアーゼ阻害剤の投与経験がある患者67例であった。24週間の治療により血漿中HIV-1 RNA量が検出限界未満(400copies/mL未満)の患者の比率は、本剤のみを投与されたHIVプロテアーゼ阻害剤の投与経験がない患者で67%(APV29005)、本剤+リトナビルが併用投与されたHIVプロテアーゼ阻害剤の投与経験がない患者でそれぞれ66%(APV20003)、70%(APV29005)、HIVプロテアーゼ阻害剤の投与経験がある患者で2試験ともに57%であった。48週間の治療により血漿中HIV-1 RNA量が検出限界未満(400copies/mL未満)の患者の比率は、HIVプロテアーゼ阻害剤の投与経験がない患者で47%、HIVプロテアーゼ阻害剤の投与経験がある患者で43%であった(APV20003での成績)(外国人データ)。24週間の治療後のCD4リンパ球数の増加量(中央値)は、本剤のみを投与されたHIVプロテアーゼ阻害剤の投与経験がない患者で353/mm3(APV29005)、本剤+リトナビルが併用投与されたHIVプロテアーゼ阻害剤の投与経験がない患者でそれぞれ127/mm3(APV20003)、131/mm3(APV29005)、HIVプロテアーゼ阻害剤の投与経験がある患者でそれぞれ114/mm3(APV20003)、149/mm3(APV29005)であった。48週間の治療後のCD4リンパ球数の増加量(中央値)は、HIVプロテアーゼ阻害剤の投与経験がない患者で163/mm3、HIVプロテアーゼ阻害剤の投与経験がある患者で145/mm3であった(APV20003での成績)(外国人データ)。
18. 薬効薬理
18.1 作用機序
ホスアンプレナビルは、アンプレナビルのプロドラッグであり、消化管上皮から吸収される過程でアンプレナビルに変換される。
アンプレナビルは、前駆体ポリ蛋白質の解裂に関与するHIVプロテアーゼを阻害することで感染性を持つウイルスの産生を抑制する5)。
本剤とリトナビルを併用投与した場合、リトナビルによる強力なCYP3A4阻害により、アンプレナビルの代謝が阻害される結果、血漿中アンプレナビル濃度が上昇する。なお、本剤とリトナビルを併用投与した場合の抗ウイルス活性は主にアンプレナビルによるものである。[16.4.2参照]
18.2 抗ウイルス活性
アンプレナビルはMT-4細胞及び末梢血白血球におけるHIV-1 IIIB及びHIV-2 ZYの複製を抑制し、IC50値はそれぞれ80nM及び340nMであった。また、末梢血白血球における臨床分離HIV-1の複製をIC50値12~19nMで抑制した。
アンプレナビルは、ジダノシン、ジドブジン、アバカビル等のヌクレオシド系逆転写酵素阻害剤(NRTI)あるいはサキナビルと併用することにより抗ウイルス活性において相乗作用を示した。また、インジナビル、リトナビルあるいはネルフィナビルと併用することにより相加作用を示した6)。
18.3 薬剤耐性
HIVをアンプレナビル存在下で培養すると耐性発現の基本となるI50Vの変異及びM46I/L、I47Vの変異がHIVプロテアーゼに生じ、これらの3変異によりアンプレナビルの抗ウイルス活性のIC50値は10倍以上上昇する7)。
また、この変異ウイルスをサキナビルの存在下で培養するとI47Vの変異が消失し、サキナビルに対する耐性を獲得すると共にアンプレナビルに対する感受性が回復した8)。一方、インジナビル、ネルフィナビルあるいはリトナビルの存在下ではそれぞれに特有の変異が出現し、2剤に耐性を示すようになる。また、in vitroではI54M、V32I+I47V及びI84Vの変異も同定されている。HIVプロテアーゼ阻害剤未治療患者において、NRTIであるアバカビル及びラミブジンの治療下でホスアンプレナビルを併用投与すると、17%(5/29例)にアンプレナビル耐性HIVが発現し、55%(16/29例)にNRTI耐性HIVが発現した(APV30001試験)。
同じくプロテアーゼ阻害剤未治療患者において、NRTI2剤の治療下でネルフィナビルを投与すると31%(17/54例)にネルフィナビル耐性HIVが発現したのに対して、ホスアンプレナビル+低用量リトナビルを併用投与してもアンプレナビル耐性HIVは発現せず(0/32例)、さらに、ホスアンプレナビル+低用量リトナビル併用時のNRTI耐性HIV発現率(13%、4/32例)もネルフィナビル投与時(57%、31/54例)に比較して低かった(APV30002試験)。
プロテアーゼ阻害剤未治療患者におけるアンプレナビル耐性HIVの発現には、I50V、I54L/M、V32I+I47VあるいはまれにI84V変異、さらにそれに続く二次変異としてのM46I/L変異が関与する可能性が示唆されている9)。
18.4 交差耐性
アンプレナビルによって発現する変異の組み合わせはアンプレナビルに特有であり、他のプロテアーゼ阻害剤ではみられない。これらの変異HIVはリトナビルに対しては多少の交差耐性を示すものの、サキナビル、インジナビル及びネルフィナビルに対する感受性は変化しない。
プロテアーゼ阻害剤既治療患者から分離したHIV中の耐性HIVの割合は、アンプレナビルが15%であり最も低かった(ロピナビル22%、サキナビル25%、インジナビル30%、リトナビル35%、ネルフィナビル55%)。また、プロテアーゼ阻害剤に対して交差耐性を示す変異HIVの74%(322/433株)がアンプレナビルに感受性を示した。アンプレナビルに対する交差耐性に関連する主な変異としてはI84V+L10I/V/Fが考えられている。
19. 有効成分に関する理化学的知見
| 一般的名称 | ホスアンプレナビルカルシウム水和物(Fosamprenavir Calcium Hydrate) |
|---|---|
| 化学名 | Monocalcium(3S)-tetrahydrofuran-3-yl(1S,2R)-3-{[(4-aminophenyl)sulfonyl](2-methylpropyl)amino}-1-benzyl-2-(phosphonatooxy)propylcarbamate hydrate |
| 分子式 | C25H34CaN3O9PS・xH2O |
| 化学構造式 | 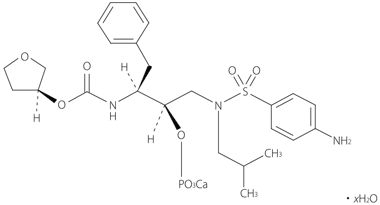 |
| 性状 | 白色~微黄白色の粉末で、水に極めて溶けにくい。 |
| 融点 | 約160℃(分解) |
22. 包装
60錠[瓶、バラ]
23. 主要文献
- DHHS:Guidelines for Using Antiretroviral Agents Among HIV-Infected Adults and Adolescents. http://www.aidsinfo.nih.gov/Guidelines/
- 抗HIV治療ガイドライン.https://hiv-guidelines.jp/
- HIV感染症「治療の手引き」.http://www.hivjp.org/
- Song I, et al.:Antimicrob Agents Chemother.2014;58(11):6696-6700
- Painter GR, et al.:Drugs of the Future.1996;21:347-350
- St.Clair MH, et al.:Antiviral Res.1996;29:53-56
- Partaledis JA, et al.:J Virol.1995;69:5228-5235
- Markland W, et al.:J Virol.2000;74:7636-7641
- Maguire M, et al.:Antimicrob Agents Chemother.2002;46:731-738
** 24. 文献請求先及び問い合わせ先
グラクソ・スミスクライン株式会社
東京都港区赤坂1-8-1
ヴィーブヘルスケア・カスタマー・サービス
TEL:0120-066-525(9:00~17:45/土日祝日及び当社休業日を除く)
https://viivhealthcare.com/ja-jp/
26. 製造販売業者等
** 26.1 製造販売元
ヴィーブヘルスケア株式会社
東京都港区赤坂1-8-1
** 26.2 販売元
グラクソ・スミスクライン株式会社
東京都港区赤坂1-8-1