テビケイ錠50mgの添付文書
- 商品名:
- テビケイ錠50mg
- 一般名:
- ドルテグラビル
- 略称 :
- DTG

添付文書の読み方
ここで提供している添付文書情報は、2025年2月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。→ PMDA
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
HIVインテグラーゼ阻害剤
ドルテグラビルナトリウム錠

- **2024年8月改訂(第7版)
- *2023年8月改訂(第6版)
規制区分:劇薬、処方箋医薬品注)
注)注意-医師等の処方箋により使用すること
| 日本標準商品分類番号 |
|---|
| 87625 |
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 5年 |
| 承認番号 | 22600AMX00561 |
| 販売開始 | 2014年4月 |
2. 禁忌(次の患者には投与しないこと)
本剤の成分に対し過敏症の既往歴のある患者
3. 組成・性状
3.1 組成
| 有効成分 | 1錠中 ドルテグラビルナトリウム52.6mg(ドルテグラビルとして50mg) |
|---|---|
| 添加剤 | D-マンニトール、結晶セルロース、ポビドン、デンプングリコール酸ナトリウム、フマル酸ステアリルナトリウム、ポリビニルアルコール(部分けん化物)、酸化チタン、マクロゴール4000、タルク、黄色三二酸化鉄 |
3.2 製剤の性状
| 識別コード | SV572 |
|---|---|
| 剤形・性状 | 黄色のフィルムコーティング錠 |
| 質量 | 309mg |
| 表 (直径) |
 約9.1mm |
| 裏 |  |
| 側面 (厚さ) |
 約4.7mm |
4. 効能又は効果
HIV感染症
5. 効能又は効果に関連する注意
本剤による治療にあたっては、患者の治療歴及び可能な場合には薬剤耐性検査(遺伝子型解析あるいは表現型解析)を参考にすること。
6. 用法及び用量
通常、成人には以下の用法・用量で経口投与する。本剤は、食事の有無にかかわらず投与できる。投与に際しては、必ず他の抗HIV薬と併用すること。
<未治療患者、インテグラーゼ阻害薬以外の抗HIV薬による治療経験のある患者>
ドルテグラビルとして50mgを1日1回経口投与する。
<インテグラーゼ阻害薬に対する耐性を有する患者>
ドルテグラビルとして50mgを1日2回経口投与する。
なお、12歳以上及び体重40kg以上の未治療、インテグラーゼ阻害薬以外の抗HIV薬による治療経験がある小児患者には、ドルテグラビルとして50mgを1日1回経口投与できる。
7. 用法及び用量に関連する注意
<未治療患者、インテグラーゼ阻害薬(INSTI)以外の抗ヒト免疫不全ウイルス(HIV)薬による治療経験のある患者>
7.1
本剤とエトラビリン(リトナビルでブーストしたアタザナビル、ダルナビル、ロピナビルと併用投与しない場合)、エファビレンツ、ネビラピン、カルバマゼピン、リファンピシン、フェニトイン、ホスフェニトイン、フェノバルビタール、セイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品を併用する場合は、本剤を50mg1日2回に増量投与すること。[10.2 参照],[16.7.3 参照]
<INSTIに対する耐性を有する患者>
7.2
本剤とエトラビリンを併用する場合は、リトナビルでブーストしたアタザナビル、ダルナビル又はロピナビルのいずれかを併用投与すること。[10.2 参照],[16.7.3 参照]
7.3
本剤とエファビレンツ、ネビラピン、ホスアンプレナビルカルシウム水和物+リトナビル、カルバマゼピン又はリファンピシンを併用しないこと。[10.2 参照],[16.7.3 参照]
8. 重要な基本的注意
8.1
本剤による治療は、抗HIV療法に十分な経験を持つ医師のもとで開始すること。
*8.2
本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又は患者に代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。
- 本剤はHIV感染症の根治療法薬ではないことから、日和見感染を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。
- 本剤は併用薬と相互作用を起こすことがあるため、服用中のすべての薬剤を担当医に報告すること。また、本剤で治療中に新たに他の薬剤を服用する場合には、事前に担当医に報告すること。
- 本剤の長期投与による影響については、現在のところ不明であること。
- 担当医の指示なしに用量を変更したり、服用を中止したりしないこと。
8.3
本剤を含む抗HIV薬の多剤併用療法を行った患者で、免疫再構築炎症反応症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染症(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
8.4
肝機能障害、黄疸があらわれることがあるので、定期的に肝機能検査を行う等、観察を十分に行うこと。[9.1.1 参照],[11.1.2 参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 B型又はC型肝炎ウイルス感染患者
肝機能の悪化(トランスアミナーゼ上昇又は増悪)のおそれがある。臨床試験において、B型又はC型肝炎ウイルス重複感染患者では、トランスアミナーゼ上昇又は増悪の発現頻度が非重複感染患者より高かった。[8.4 参照],[11.1.2 参照]
**9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
海外の観察研究において、無脳症や二分脊椎などの神経管閉鎖障害が、受胎前からドルテグラビル含有製剤を服用していた妊婦から生まれた児9460例中10例(0.11%、95%信頼区間0.06-0.19)に報告されており、ドルテグラビルを含まない抗HIV薬を服用していた妊婦から生まれた児23664例中25例(0.11%、95%信頼区間0.07-0.16)、HIV陰性の妊婦から生まれた児170723例中108例(0.07%、95%信頼区間0.05-0.08)に報告されている1)。
ドルテグラビルはヒト胎盤を通過する。ドルテグラビルの母体血漿中濃度に対する胎児臍帯血漿中濃度の比(中央値[範囲])は、1.28[1.21-1.28]であることが報告されている2)(外国人データ)。
9.6 授乳婦
授乳を避けさせること。一般に、乳児へのHIV感染を防ぐため、あらゆる状況下においてHIVに感染した女性は授乳すべきでない。 ドルテグラビルはヒト乳汁中に移行する。ドルテグラビルの母体血漿中濃度に対する乳汁中濃度の比(中央値[範囲])は、0.033[0.021-0.050]であることが報告されている2)(外国人データ)。
9.7 小児等
低出生体重児、新生児、乳児、幼児又は12歳未満又は体重40kg未満の小児を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら注意して投与すること。一般に生理機能(肝機能、腎機能、心機能等)が低下しており、合併症を有している又は他の薬剤を併用している場合が多い。
10. 相互作用
本剤は主にUGT1A1の基質であり、一部CYP3A4でも代謝される。また、本剤は有機カチオントランスポーター2(OCT2)及びMultidrug and Toxin Extrusion 1(MATE1)を阻害する。[16.4.1 参照],[16.7.1 参照]
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| ピルシカイニド塩酸塩水和物 | ピルシカイニドの血漿中濃度を増加させる可能性がある。併用により、ピルシカイニドで重大な副作用として報告されている心室頻拍、洞停止及び心室細動等の発現及び重篤化があらわれるおそれがあるので、併用中は注意深く観察すること。 | 本剤のOCT2及びMATE1の阻害作用により、ピルシカイニドの排出が阻害される可能性がある。 |
| エトラビリン [7.1 参照],[7.2 参照],[16.7.3 参照] |
本剤の血漿中濃度をCmaxで52%、Cτで88%低下させたとの報告がある3)。 | これらの薬剤がCYP3A4及びUGT1A1を誘導することにより、本剤の代謝が促進される。 |
| エファビレンツ [7.1 参照],[7.3 参照],[16.7.3 参照] |
本剤の血漿中濃度をCmaxで39%、Cτで75%低下させたとの報告がある4)。 | |
| ネビラピン [7.1 参照],[7.3 参照] |
本剤の血漿中濃度を低下させる可能性がある。 | |
| ホスアンプレナビルカルシウム水和物+リトナビル [7.3 参照],[16.7.3 参照] |
本剤の血漿中濃度をCmaxで24%、Cτで49%低下させたとの報告がある5)ため、INSTIに対する耐性を有する患者では、本剤と併用しないこと。 | ホスアンプレナビルがCYP3A4及びUGT1A1を誘導することにより、本剤の代謝が促進される。 |
| カルバマゼピン [7.1 参照],[7.3 参照],[16.7.3 参照] |
本剤の血漿中濃度をCmaxで33%、Cτで73%低下させたとの報告がある6)。 | カルバマゼピンがCYP3A4及びUGT1A1を誘導することにより、本剤の代謝が促進される。 |
|
本剤の血漿中濃度を低下させる可能性がある。 | これらの薬剤並びにセイヨウオトギリソウがCYP3A4及びUGT1A1を誘導することにより、本剤の代謝が促進される。 |
| リファンピシン [7.1 参照],[7.3 参照],[16.7.3 参照] |
本剤の血漿中濃度をCmaxで43%、Cτで72%低下させたとの報告がある7)。 | リファンピシンがCYP3A4及びUGT1A1を誘導することにより、本剤の代謝が促進される。 |
| 多価カチオン(Mg, Al等)含有製剤 [16.7.3 参照] |
本剤の血漿中濃度をCmaxで72%、C24で74%低下させる8)。本剤は多価カチオン含有制酸剤の投与2時間前又は6時間後の投与が推奨される。 | これらの多価カチオンと錯体を形成することにより、本剤の吸収が阻害される。 |
| 鉄剤、カルシウム含有製剤(サプリメント等) [16.7.3 参照] |
本剤の血漿中濃度をCmaxで35%、C24で32%低下させる8)。食事と同時に摂取する場合を除き、本剤は鉄剤、カルシウム含有製剤の投与2時間前又は6時間後の投与が推奨される。 | 鉄、カルシウムと錯体を形成することにより、本剤の吸収が阻害される。 |
| メトホルミン塩酸塩 [16.7.2 参照] |
メトホルミンの血漿中濃度をドルテグラビル50mg 1日1回投与時及び1日2回投与時にCmaxでそれぞれ66%及び111%上昇させる9)。注意深く観察し、必要に応じてメトホルミンを減量する等慎重に投与すること。 | 本剤のOCT2及びMATE1の阻害作用により、メトホルミンの排出が阻害される可能性がある。 |
11. 副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1 重大な副作用
11.1.1 薬剤性過敏症症候群(1%未満)
初期症状として発疹、発熱がみられ、さらに肝機能障害、リンパ節腫脹、好酸球増多等を伴う遅発性の重篤な過敏症状があらわれることがある。なお、投与中止後も発疹、発熱、肝機能障害等の症状が再燃あるいは遷延化することがあるので注意すること。
11.1.2 肝機能障害、黄疸(いずれも1%未満)
AST、ALT、ビリルビンの上昇等を伴う肝機能障害、黄疸があらわれることがある。[8.4 参照],[9.1.1 参照]
11.2 その他の副作用
| 2%以上 | 1~2%未満 | 1%未満 | 頻度不明 | |
|---|---|---|---|---|
| 免疫系 | 免疫再構築炎症反応症候群 | |||
| 精神・神経系 | 頭痛、不眠症、めまい、異常な夢 | うつ病、不安 | 自殺念慮、自殺企図 | |
| 消化器 | 悪心、下痢、嘔吐 | 上腹部痛、鼓腸 | 腹部不快感、腹痛 | |
| 肝臓 | 肝炎 | |||
| 皮膚 | 発疹、そう痒 | |||
| 全身症状 | 疲労 | |||
| 筋骨格 | 関節痛、筋肉痛 | |||
| 臨床検査 | ビリルビン上昇、クレアチニン上昇、体重増加 | CK上昇 |
13. 過量投与
13.1 処置
血液透析により除去される可能性は低いことが報告されている10),11)。
16. 薬物動態
16.1 血中濃度
16.1.1 単回経口投与
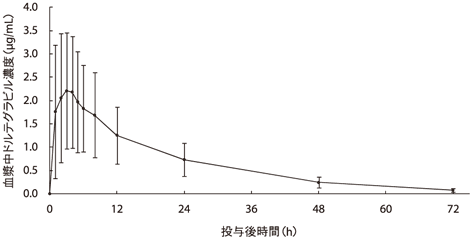
健康成人男性6例及び女性4例に本剤50mgを単回経口投与した時の血漿中ドルテグラビル濃度推移を図-1に、ドルテグラビルの薬物動態パラメータを表-1に示す。ドルテグラビルは投与後約3時間で最高血漿中濃度に達し、消失半減期は約15時間であった。また、日本人における薬物動態は外国人における薬物動態と同様であった12)。
| Cmax (μg/mL) |
Tmax注1) (h) |
AUC0-inf (μg・h/mL) |
t1/2 (h) |
C24 (μg/mL) |
|---|---|---|---|---|
| 2.37±1.23 | 3.0(2.0-4.0) | 47.7±24.6 | 14.7±1.56 | 0.73±0.36 |
平均値±標準偏差、10例
注1)中央値(範囲)
本剤は経口投与により速やかに吸収され、投与後約2~3時間で最高血漿中濃度に達した。HIV感染症患者及び健康成人に本剤を経口投与した時の血漿中ドルテグラビルの曝露量は、2~100mg注)の範囲では投与量増加の割合を下回って増加した13),14)が、25~50mg注)の範囲では投与量にほぼ比例して増加した15)(外国人データ)。
16.1.2 反復経口投与
成人HIV感染症患者における後期第II相及び第III相試験の母集団薬物動態解析で推定した定常状態におけるドルテグラビルの薬物動態パラメータを表-2に示す(外国人データ)。
| パラメータ | AUC0-24(μg・h/mL) | Cmax(μg/mL) | Cτ(μg/mL) |
|---|---|---|---|
| 50mg 1日1回 | 53.6(27) | 3.67(20) | 1.11(46) |
| 50mg 1日2回 | 75.1(35) | 4.15(29) | 2.12(47) |
母集団薬物動態解析に基づく推定値
幾何平均値(CV%)
16.1.3 小児等
抗HIV薬による治療経験のある小児HIV感染症患者(12~18歳未満、10例)に本剤50mgを1日1回経口投与した時の薬物動態は成人と同様であった。小児患者での血漿中ドルテグラビルの薬物動態パラメータを表-3に示す(外国人データ)。
| 年齢/体重 | 用量 | 薬物動態パラメータの推定値 | ||
|---|---|---|---|---|
| AUC0-24 (μg・h/mL) |
Cmax (μg/mL) |
C24 (μg/mL) |
||
| 12歳以上18歳未満 体重40kg以上注1) |
50mg注1) 1日1回 |
46 (43) |
3.49 (38) |
0.90 (59) |
幾何平均値(CV%)、10例
注1)体重が37kgであった1例には35mg注)を1日1回投与した。
16.2 吸収
16.2.1 食事の影響
本剤は食事の有無にかかわらず投与できる。健康成人に対し、低、中又は高脂肪食(それぞれ7%脂肪/300kcal、30%脂肪/600kcal又は53%脂肪/870kcal)を摂取後に本剤50mgを単回経口投与した場合、血漿中ドルテグラビルのAUC0-infは絶食下と比較してそれぞれ33、41及び66%増加し、Cmaxはそれぞれ46、52及び67%増加した。また、Tmaxはそれぞれ3、4及び5時間であり、食事によりドルテグラビルの吸収量は増加し、吸収速度が低下した(外国人データ)。
16.3 分布
16.3.1 血漿蛋白結合率
In vitroにおいて、ドルテグラビルのヒト血漿蛋白結合率は約99.3%であった16)。
16.3.2 分布容積
健康成人男性にドルテグラビル20mg(懸濁液)注)を単回経口投与した時の見かけの分布容積は12.5Lであった(外国人データ)。
16.3.3 血球移行性
ヒトでの血液/血漿比(平均値)は0.441~0.535であり、ドルテグラビルの血球移行性は低かった(5%未満)。
16.3.4 非結合型薬物
血漿中ドルテグラビルの遊離分画は健康成人で約0.2~1.1%、中等度の肝機能障害患者で約0.4~0.5%、重度の腎機能障害患者で約0.8~1.0%、HIV感染症患者で0.5%であった(外国人データ)。
16.3.5 脳脊髄液への移行
ドルテグラビルは脳脊髄液中にも分布する。本剤50mg及びアバカビル・ラミブジン(600mg・300mg)が併用投与された抗HIV薬による治療経験のない成人HIV感染症患者11例において、ドルテグラビルの脳脊髄液中濃度(中央値)は18ng/mLであり、血漿中濃度の0.11~0.66%であった(外国人データ)。
16.3.6 組織内分布
ドルテグラビルは女性及び男性の生殖器に分布する。
健康成人女性に本剤50mg/日を5~7日間経口投与した時の子宮頸膣液、子宮頸部組織及び膣組織におけるドルテグラビルのAUCは定常状態での血漿中ドルテグラビルのAUCの6~10%であった(外国人データ)。
また、健康成人男性に本剤50mg/日を8日間経口投与した時の精液及び直腸組織におけるドルテグラビルAUCは定常状態での血漿中ドルテグラビルのAUCの7及び17%であった(外国人データ)。
16.4 代謝
16.4.1 主な代謝酵素
In vitroにおいて、ドルテグラビルは主に肝臓のUGT1A1でグルクロン酸抱合される17)。また、ドルテグラビルはCYP3Aでも一部代謝された18)。[10. 参照]
16.5 排泄
健康成人に14C-ドルテグラビル20mg(懸濁液)注)を単回経口投与した時の総投与量の約9.7%が酸化的代謝物として尿糞中に回収された(外国人データ)。
健康成人にドルテグラビル20mg注)を単回経口投与した時の主な排泄経路は糞であり、経口投与量の53%が未変化体として糞中に排泄された。また、尿中には経口投与量の31%が排泄され、その内訳は18.9%がエーテル型グルクロン酸抱合体、3.6%がN-脱アルキル体、3.0%がベンジル位の酸化体であり、未変化体は1%未満であった(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
重度の腎機能障害(8例、クレアチニンクリアランス(Ccr):30mL/min未満)を有する患者に本剤50mgを単回経口投与した時の血漿中ドルテグラビルの薬物動態パラメータを表-4に示す(外国人データ)。重度の腎機能障害患者における薬物動態は健康成人との間に臨床的に重要である差はみられなかったことから、腎機能障害患者に対して本剤の用量調節を行う必要はない19)。
| 被験者 | Cmax (μg/mL) |
AUC0-inf (μg・h/mL) |
t1/2 (h) |
|---|---|---|---|
| 重度の腎機能障害患者 | 1.50(34) | 23.5(48) | 12.7(31) |
| 健康成人 | 1.86(45) | 37.1(58) | 15.4(15) |
幾何平均値(CV%)
16.6.2 肝機能障害患者
ドルテグラビルは主に肝臓で代謝されて排泄される。中等度の肝機能障害(8例、Child-Pugh分類:B)を有する患者に本剤50mgを単回経口投与した時の血漿中ドルテグラビルの薬物動態パラメータを表-5に示す(外国人データ)。中等度の肝機能障害患者における薬物動態は健康成人と同様であったことから、中等度の肝機能障害に対して本剤の用量調節の必要はない20)。なお、重度の肝機能障害患者での本剤の薬物動態に及ぼす影響については検討していない。
| 被験者 | AUC0-inf (μg・h/mL) |
Cmax (μg/mL) |
C24 (μg/mL) |
|---|---|---|---|
| 中等度の肝機能障害患者 | 38.5(30) | 1.78(17) | 0.59(36) |
| 健康成人 | 37.3(47) | 1.80(49) | 0.57(44) |
幾何平均値(CV%)
16.6.3 B型肝炎及びC型肝炎のウイルス重複感染患者
C型肝炎ウイルス重複感染患者を対象とした母集団薬物動態解析の結果、C型肝炎ウイルス重複感染はドルテグラビルの曝露量に対して臨床的な影響を及ぼさなかった(外国人データ)。なお、B型肝炎ウイルス重複感染患者における本剤投与時の薬物動態データは限られている。
16.6.4 性別
健康成人にドルテグラビル250mg(懸濁液)注)を単回経口投与した時の血漿中ドルテグラビルの薬物動態パラメータは、男性(17例)よりも女性(24例)の方がわずか(最大約20%)に高い傾向がみられた(外国人データ)。
成人HIV感染症患者を対象とした後期第II相及び第III相試験での母集団薬物動態解析の結果、性別はドルテグラビルの曝露量に対して臨床的な影響を及ぼさなかった(外国人データ)。
16.6.5 人種
成人HIV感染症患者を対象とした後期第II相及び第III相試験での母集団薬物動態解析の結果、人種はドルテグラビルの曝露量に対して臨床的な影響を及ぼさなかった(外国人データ)。
16.7 薬物相互作用
16.7.1 In vitro試験
(1) 分布に関わるトランスポーター
ドルテグラビルはヒトPgp及びBCRPの基質である21),22)。
(2) 排泄に関わるトランスポーター
ドルテグラビルはヒト有機アニオントランスポーター1(OAT1)、OAT3、OCT2、MATE1及びMATE2-Kを介した輸送を阻害した(IC50:それぞれ2.12、1.97、1.93、6.34及び24.8μM)23),24)。[10. 参照]
16.7.2 本剤が併用薬の薬物動態に及ぼす影響
ドルテグラビルが併用薬の薬物動態に及ぼす影響を、表-6に示す(外国人データ)。[10.2 参照]
| 併用薬及び用量 | ドルテグラビル |
例数 | ドルテグラビル併用時/非併用時の併用薬の 薬物動態パラメータの幾何平均比 (90%信頼区間);影響なし=1.00 |
||
|---|---|---|---|---|---|
| Cτ又はC24 | AUC | Cmax | |||
| エチニルエストラジオール 0.035mg |
50mg 1日2回 |
15 | 1.02 (0.93, 1.11) |
1.03 (0.96, 1.11) |
0.99 (0.91, 1.08) |
| メサドン 20-150mg | 50mg 1日2回 |
11 | 0.99 (0.91, 1.07) |
0.98 (0.91, 1.06) |
1.00 (0.94, 1.06) |
| ミダゾラム 3mg | 25mg 1日1回 |
10 | - | 0.95 (0.79, 1.15) |
- |
| Norelgestromin (国内未発売) 0.25mg |
50mg 1日2回 |
15 | 0.93 (0.85, 1.03) |
0.98 (0.91, 1.04) |
0.89 (0.82, 0.97) |
| リルピビリン 25mg 1日1回 |
50mg 1日1回 |
16 | 1.21 (1.07, 1.38) |
1.06 (0.98, 1.16) |
1.10 (0.99, 1.22) |
| テノホビル ジソプロキシルフマル酸塩 300mg 1日1回 |
50mg 1日1回 |
15 | 1.19 (1.04, 1.35) |
1.12 (1.01, 1.24) |
1.09 (0.97, 1.23) |
| メトホルミン 500mg 1日2回 |
50mg 1日1回 |
14 | - | 1.79 (1.65, 1.93) |
1.66 (1.53, 1.81) |
| メトホルミン 500mg 1日2回 |
50mg 1日2回 |
14 | - | 2.45 (2.25, 2.66) |
2.11 (1.91, 2.33) |
16.7.3 併用薬が本剤の薬物動態に及ぼす影響
併用薬がドルテグラビルの薬物動態に及ぼす影響を、表-7に示す(外国人データ)。[7.1 参照],[7.2 参照],[7.3 参照],[10.2 参照]
| 併用薬及び用量 | ドルテグラビル |
例数 | 他剤併用時/非併用時のドルテグラビルの 薬物動態パラメータの 幾何平均比(90%信頼区間);影響なし=1.00 |
||
|---|---|---|---|---|---|
| Cτ又はC24 | AUC | Cmax | |||
| アタザナビル 400mg 1日1回 |
30mg 1日1回 |
12 | 2.80 (2.52, 3.11) |
1.91 (1.80, 2.03) |
1.50 (1.40, 1.59) |
| アタザナビル+リトナビル 300mg+100mg 1日1回 |
30mg 1日1回 |
12 | 2.21 (1.97, 2.47) |
1.62 (1.50, 1.74) |
1.34 (1.25, 1.42) |
| テノホビル ジソプロキシルフマル酸塩 300mg 1日1回 |
50mg 1日1回 |
15 | 0.92 (0.82, 1.04) |
1.01 (0.91, 1.11) |
0.97 (0.87, 1.08) |
| ダルナビル+リトナビル 600mg+100mg 1日2回 |
30mg 1日1回 |
15 | 0.62 (0.56, 0.69) |
0.78 (0.72, 0.85) |
0.89 (0.83, 0.97) |
| エファビレンツ 600mg 1日1回 |
50mg 1日1回 |
12 | 0.25 (0.18, 0.34) |
0.43 (0.35, 0.54) |
0.61 (0.51, 0.73) |
| エトラビリン 200mg 1日2回 |
50mg 1日1回 |
15 | 0.12 (0.09, 0.16) |
0.29 (0.26, 0.34) |
0.48 (0.43, 0.54) |
| エトラビリン+ダルナビル+リトナビル 200mg+600mg+100mg 1日2回 |
50mg 1日1回 |
9 | 0.63 (0.52, 0.76) |
0.75 (0.69, 0.81) |
0.88 (0.78, 1.00) |
| ホスアンプレナビル+リトナビル 700mg+100mg 1日2回 |
50mg 1日1回 |
12 | 0.51 (0.41, 0.63) |
0.65 (0.54, 0.78) |
0.76 (0.63, 0.92) |
| ロピナビル・リトナビル 400mg・100mg 1日2回 |
30mg 1日1回 |
15 | 0.94 (0.85, 1.05) |
0.97 (0.91, 1.04) |
1.00 (0.94, 1.07) |
| 乾燥水酸化アルミニウムゲル・水酸化マグネシウム 20mL 単回 |
50mg 単回 |
16 | 0.26 (0.21, 0.31) |
0.26 (0.22, 0.32) |
0.28 (0.23, 0.33) |
| 乾燥水酸化アルミニウムゲル・水酸化マグネシウム 20mL 本剤投与2時間後 単回 |
50mg 単回 |
16 | 0.70 (0.58, 0.85) |
0.74 (0.62, 0.90) |
0.82 (0.69, 0.98) |
| 総合ビタミン剤 1錠 1日1回 |
50mg 単回 |
16 | 0.68 (0.56, 0.82) |
0.67 (0.55, 0.81) |
0.65 (0.54, 0.77) |
| オメプラゾール 40mg 1日1回 |
50mg 単回 |
12 | 0.95 (0.75, 1.21) |
0.97 (0.78, 1.20) |
0.92 (0.75, 1.11) |
| prednisone(国内未発売) 60mg 1日1回(漸減) |
50mg 1日1回 |
12 | 1.17 (1.06, 1.28) |
1.11 (1.03, 1.20) |
1.06 (0.99, 1.14) |
| リファンピシン注1) 600mg 1日1回 |
50mg 1日2回注1) |
11 | 0.28 (0.23, 0.34) |
0.46 (0.38, 0.55) |
0.57 (0.49, 0.65) |
| リファンピシン注2) 600mg 1日1回 |
50mg 1日2回注2) |
11 | 1.22 (1.01, 1.48) |
1.33 (1.15, 1.53) |
1.18 (1.03, 1.37) |
| リファブチン 300mg 1日1回 |
50mg 1日1回 |
9 | 0.70 (0.57, 0.87) |
0.95 (0.82, 1.10) |
1.16 (0.98, 1.37) |
| リルピビリン 25mg 1日1回 |
50mg 1日1回 |
16 | 1.22 (1.15, 1.30) |
1.12 (1.05, 1.19) |
1.13 (1.06, 1.21) |
| Tipranavir(国内未発売)+リトナビル 500mg+200mg 1日2回 |
50mg 1日1回 |
14 | 0.24 (0.21, 0.27) |
0.41 (0.38, 0.44) |
0.54 (0.50, 0.57) |
| テラプレビル 750mg 8時間ごと |
50mg 1日1回 |
15 | 1.37 (1.29, 1.45) |
1.25 (1.20, 1.31) |
1.19 (1.11, 1.26) |
| Boceprevir(国内未発売) 800mg 8時間ごと |
50mg 1日1回 |
13 | 1.08 (0.91, 1.28) |
1.07 (0.95, 1.20) |
1.05 (0.96, 1.15) |
| カルバマゼピン 300mg 1日2回 |
50mg 1日1回 |
14 | 0.27 (0.24, 0.31) |
0.51 (0.48, 0.55) |
0.67 (0.61, 0.73) |
注1)ドルテグラビル50mg 1日2回投与とリファンピシンを併用したドルテグラビル50mg 1日2回投与との比較
注2)ドルテグラビル50mg 1日1回投与とリファンピシンを併用したドルテグラビル50mg 1日2回投与との比較
注)本剤の承認された用法及び用量は、「未治療患者、INSTI以外の抗HIV薬による治療経験のある患者は、ドルテグラビルとして50mgを1日1回経口投与する。INSTIに対する耐性を有する患者は、ドルテグラビルとして50mgを1日2回経口投与する。なお、12歳以上及び体重40kg以上の未治療、INSTI以外の抗HIV薬による治療経験がある小児患者には、ドルテグラビルとして50mgを1日1回経口投与できる。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第III相試験(SPRING-2:ING113086)
抗HIV薬による治療経験のない成人HIV感染症患者822例を対象とした二重盲検比較試験において、ドルテグラビル50mgを1日1回投与した群(ドルテグラビル投与群)と、ラルテグラビル400mgを1日2回投与した群(ラルテグラビル投与群)に、それぞれ411例の患者が無作為に割り付けられた。その結果、主要評価項目である投与48週後のHIV-1 RNA量が50copies/mL未満であった患者の割合は、ラルテグラビル投与群の85%に対して、ドルテグラビル投与群は88%であり、群間差(95%信頼区間)は、2.5%(-2.2%, 7.1%)であり、ラルテグラビルに対するドルテグラビルの非劣性が示された(非劣性マージン10%)。投与96週後のHIV-1 RNA量が50copies/mL未満であった患者の割合は、ラルテグラビル投与群の76%に対して、ドルテグラビル投与群は81%であった25),26)。
副作用発現頻度は、ドルテグラビル投与群で28%(116/411例)であった。主な副作用は、悪心10%(40/411例)及び頭痛4%(18/411例)であった。(投与48週時)。
なお、本試験における試験成績の要約を表-1に示した。
| 結果 | ドルテグラビル50mg 1日1回 + ヌクレオシド系逆転写 酵素阻害剤2剤 (411例) |
ラルテグラビル400mg 1日2回 + ヌクレオシド系逆転写 酵素阻害剤2剤 (411例) |
||
|---|---|---|---|---|
| 48週 | 96週 | 48週 | 96週 | |
| HIV-1 RNA量が 50copies/mL未満 |
361例 (88%) |
332例 (81%) |
351例 (85%) |
314例 (76%) |
| 両群間の差注1) (95%信頼区間) |
2.5% (-2.2%, 7.1%) |
4.5% (-1.1%, 10.0%) |
- | |
| ウイルス学的な 治療失敗注2) |
20例 (5%) |
22例 (5%) |
31例 (8%) |
43例 (10%) |
注1)ベースラインの層別因子により調整
注2)ウイルス学的効果が不十分のため、投与48週又は96週後までに背景療法の組合せを変更又は試験薬剤の投与を中止した症例、若しくは48週又は96週目にHIV-1 RNA量が50copies/mL以上であった症例
17.1.2 海外第III相試験(SINGLE:ING114467)
抗HIV薬による治療経験のない成人HIV感染症患者833例を対象とした二重盲検比較試験において、ドルテグラビル50mg(1日1回投与)とアバカビル・ラミブジンによる併用投与群(ドルテグラビル投与群)に414例、エファビレンツ・テノホビル・エムトリシタビン投与群(対照群)に419例が無作為に割り付けられた。その結果、主要評価項目である投与48週後のHIV-1 RNA量が50copies/mL未満であった患者の割合は、対照群の81%に対して、ドルテグラビル投与群は88%であり、群間差(95%信頼区間)は、7.4%(2.5%, 12.3%)であり、対照に対するドルテグラビルの非劣性が示された(非劣性マージン10%)。また、投与96週後のHIV-1 RNA量が50copies/mL未満であった患者の割合は、対照群の72%に対して、ドルテグラビル投与群は80%であった。さらに、投与96週後以降に非盲検下で継続投与を行った結果、144週後のHIV-1 RNA量が50copies/mL未満であった患者の割合は、ドルテグラビル投与群では71%、対照群では63%であった。ウイルス学的な治療失敗は、ドルテグラビル投与群の10%及び対照群の7%で認められた27)。
副作用発現頻度は、ドルテグラビル投与群で43%(180/414例)であった。主な副作用は、悪心10%(42/414例)、不眠10%(41/414例)及び浮動性めまい7%(28/414例)であった(投与48週時)。
なお、本試験における試験成績の要約を表-2に示した。
| 結果 | ドルテグラビル50mg 1日1回 + アバカビル・ラミブジン注1) (414例) |
エファビレンツ・ 1日1回 (419例) |
||||
|---|---|---|---|---|---|---|
| 48週 | 96週 | 144週 | 48週 | 96週 | 144週 | |
| HIV-1 RNA量が 50copies/mL未満 |
364例 (88%) |
332例 (80%) |
296例 (71%) |
338例 (81%) |
303例 (72%) |
265例 (63%) |
| 両群間の差注3) (95%信頼区間) |
7.4% (2.5%, 12.3%) |
8.0% (2.3%, 13.8%) |
8.3% (2.0%, 14.6%) |
- | ||
| ウイルス学的な 治療失敗注4) |
21例 (5%) |
31例 (7%) |
43例 (10%) |
26例 (6%) |
33例 (8%) |
30例 (7%) |
注1)アバカビル600mg・ラミブジン300mgをエプジコム®配合錠として1日1回投与
注2)エファビレンツ600mg・テノホビルジソプロキシルフマル酸塩300mg・エムトリシタビン200mgをAtripla®配合錠として1日1回投与
注3)ベースラインの層別因子により調整
注4)ウイルス学的効果が不十分のため、投与48週、96週又は144週後までに試験薬剤の投与を中止した症例、若しくは48週、96週又は144週目にHIV-1 RNA量が50copies/mL以上であった症例
17.1.3 海外第III相試験(SAILING:ING111762)
抗HIV薬による治療経験があり、かつINSTIの投与経験のない成人HIV感染症患者715例を対象とした二重盲検比較試験において、背景療法とドルテグラビル50mg 1日1回投与を併用した群(ドルテグラビル投与群)と、背景療法とラルテグラビル400mg 1日2回投与を併用した群(ラルテグラビル投与群)に、それぞれ354例及び361例の患者が無作為に割り付けられた。その結果、主要評価項目である投与48週後のHIV-1 RNA量が50copies/mL未満であった患者の割合は、ラルテグラビル投与群の64%に対して、ドルテグラビル投与群は71%であり、群間差(95%信頼区間)は、7.4%(0.7%, 14.2%)であり、ラルテグラビルに対するドルテグラビルの非劣性が示された(非劣性マージン12%)28)。
副作用発現頻度は、ドルテグラビル投与群で20%(73/357例)であった。主な副作用は、下痢8%(29/357例)及び悪心4%(13/357例)であった(投与48週時)。
なお、本試験における試験成績の要約を表-3に示した。
| 結果 | ドルテグラビル50mg 1日1回 + 背景療法 (354例)注1) |
ラルテグラビル400mg 1日2回 + 背景療法 (361例)注1) |
|---|---|---|
| 48週 | 48週 | |
| HIV-1 RNA量が 50copies/mL未満 |
251例(71%) | 230例(64%) |
| 両群間の差注2) (95%信頼区間) |
7.4% (0.7%, 14.2%) |
|
| ウイルス学的な治療失敗 | 71例(20%) | 100例(28%) |
注1)1実施施設において、データ整合性のため4例が有効性解析から除外
注2)ベースラインの層別因子により調整
17.1.4 海外第III相試験(VIKING-3:ING112574)
INSTIに耐性を有する成人HIV感染症患者183例を対象とした非盲検非対照試験において、ドルテグラビル50mg 1日2回投与による有効性及び安全性を検討した。ドルテグラビルと併用する背景療法は、投与7日目までは試験開始前からの治療法を継続し、8日目以降は最適な背景療法を行った。対象患者183例のうち133例で試験開始時にINSTIに対する耐性変異が認められた。その他の50例には試験開始前にINSTIに対する耐性を示す治療歴はあったが、試験開始時には耐性が確認されなかった。試験開始時から投与8日目までのHIV-1 RNAの変化量(平均値)は、-1.4 log10 copies/mL(95%信頼区間:-1.5~-1.3 log10 copies/mL)であり、試験開始時と比較して有意に減少した(p<0.001)。INSTIに対する耐性変異毎のウイルス学的効果を表-4に示す29)。
| INSTIに対する 耐性変異 |
症例数 | HIV-1 RNAの変化量 (log10 copies/mL) 平均(標準偏差) |
HIV-1 RNA量が 1.0 log10以上減少 した症例の割合注1) |
|---|---|---|---|
| Q148H/K/R 変異なし注2) |
124 | -1.60(0.52) | 114(92%) |
| Q148及び 二次変異注3) 1ヵ所 |
35 | -1.18(0.52) | 25(71%) |
| Q148及び 二次変異注3) 2ヵ所以上 |
20 | -0.92(0.81) | 9(45%) |
注1)投与8日目にHIV-1 RNA量が50copies/mL未満であった症例を含む
注2)INSTIに対する耐性変異(N155H, Y143C/H/R, T66A, E92Q)若しくは試験開始前からINSTIに対する耐性を示す治療歴のみがあった場合
注3)G140A/C/S, E138A/K/T, L74I
データカットオフ時には、組み入れ症例(183例)の全例が投与後24週を経過していた。主要評価項目である投与24週後のHIV-1 RNA量が50copies/mL未満であった患者の割合は、183例中126例(69%)であった。INSTIに対する耐性変異毎のウイルス学的効果を表-5に示す。投与24週後のHIV-1 RNA量が50copies/mL未満であった患者の割合が最も少なかったのは、Q148変異に加えて2ヵ所以上の変異をもつ患者であった。
副作用発現頻度は、ドルテグラビル投与群で25%(45/183例)であった。主な副作用は、悪心5%(10/183例)、下痢5%(10/183例)及び頭痛5%(9/183例)であった(投与24週時)。
| INSTIに対する耐性変異 | HIV-1 RNA量が |
|---|---|
| Q148H/K/R変異なし注1) | 96/114(84%) |
| Q148及び二次変異注2)1ヵ所 | 20/31(65%) |
| Q148及び二次変異注2)2ヵ所以上 | 4/16(25%) |
注1)INSTIに対する耐性変異(N155H, Y143C/H/R, T66A, E92Q)若しくは試験開始前からINSTIに対する耐性を示す治療歴のみあった場合
注2)G140A/C/S, E138A/K/T, L74I
18. 薬効薬理
18.1 作用機序
ドルテグラビルはレトロウイルスの複製に必要な酵素であるHIVインテグラーゼの活性部位に結合することによってその活性を阻害し、ウイルスDNAの宿主DNAへの組込みを抑制する。
18.2 抗ウイルス作用
HIV-1 BaL株及びHIV-1 NL432株に感染させた末梢血単核球を用いた時のドルテグラビルのウイルス複製に対する50%阻害濃度(IC50)は、それぞれ0.51及び0.53nMであり、HIV-1 IIIB株に感染させたMT-4細胞を用いた時のIC50は2.1nMであった(in vitro)。
13種のHIV-1臨床分離株(サブタイプB)のインテグラーゼコード領域を導入した組換えウイルスに対するドルテグラビルのIC50(平均値)は0.52nMであり、その活性は実験室株に対する抗ウイルス活性と同程度であった。24種のHIV-1臨床分離株[グループM(サブタイプA、B、C、D、E、F、G)及びグループO]並びに3種のHIV-2臨床分離株からなるパネル株を感染させた末梢血単核球を用いた時のドルテグラビルのIC50(幾何平均)はHIV-1株及びHIV-2株でそれぞれ0.20nM(範囲は0.02~2.14nM)及び0.18nM(範囲は0.09~0.61nM)であった(in vitro)。
18.3 薬剤耐性
18.3.1 臨床試験成績
抗HIV薬による治療経験があり、かつINSTIの投与経験のない患者を対象としたSAILING試験(ドルテグラビル投与群354例)において、投与48週後にウイルス学的な治療失敗例の17例中4例でINSTIに耐性が認められた。これら4例中2例に特有のR263Kインテグラーゼ変異が認められ、FCの最大値は1.93であった。もう1例には、多型のV151V/Iインテグラーゼ変異が認められFCの最大値は0.92であり、残り1例には試験前からインテグラーゼ変異の存在が認められており、既にINSTIの投与経験があるか、又はインテグラーゼ耐性ウイルスに感染したものと推定された。
18.3.2 In vitro試験
HIV-1 IIIB株及びHIV-1 NL432株をそれぞれ112及び56日間継代培養した試験でみられたインテグラーゼ領域のアミノ酸変異はS153Y、S153F、E92Q及びG193Eであり、FC(各種分離株に対するIC50/野生型HIV-1株に対するIC50)の最大値は4.1であった。また、HIV-1 臨床分離株(サブタイプB、C及びA/G)を更に長期間継代培養した試験でみられた変異はG118R(FC=10)、S153T及びR263K(FC=1.5)であった。18.4 交差耐性
18.4.1 In vitro試験
ラルテグラビル[Fold Change(FC)>81]に対する遺伝子型及び表現型の耐性を有する30種の臨床分離株について、ドルテグラビル(FC=1.5)に対する感受性を調べた。G140S+Q148H分離株では、ドルテグラビルのFC値は3.75であり、G140S+Q148R分離株では13.3、T97A+Y143R分離株では1.05、N155H分離株では1.37であった。ラルテグラビルの投与経験のある患者から分離した705種のラルテグラビル耐性株について、ドルテグラビルに対する感受性を調べたところ、93.9%の分離株に対してFCが10以下であった。
部位特異的変異を有する60種のINSTI耐性HIV-1ウイルスパネル株(28種は単一アミノ酸変異、32種は二重又は多重変異)を用いてドルテグラビルの抗ウイルス活性を検討した。単一のINSTI耐性関連アミノ酸変異(T66K、I151L及びS153Y)を有するウイルスでは、ドルテグラビルに対する感受性が2倍以上(2.3~3.6倍)低下した。複数の変異(T66K/L74M、E92Q/N155H、G140C/Q148R、G140S/Q148H、G140S/Q148R、G140S/Q148K、Q148R/N155H、T97A/G140S/Q148、及びE138/G140/Q148)を有するウイルスでは、ドルテグラビルに対する感受性が2倍以上(2.5~21倍)低下した。
18.4.2 臨床試験成績
INSTIに耐性を有する患者を対象としたVIKING-3試験では、投与24週後までに183例中36例でウイルス学的な治療失敗が認められた。このうち31例については、試験開始時及びウイルス学的な治療失敗時の両時点で解析用耐性データがあり、31例中16例(52%)で投与に伴う変異が認められた。確認された治療下での変異又は混合変異はL74L/M(1例)、E92Q(2例)、T97A(8例)、E138K/A(7例)、G140S(2例)、Y143H(1例)、S147G(1例)、Q148H/K/R(4例)、N155H(1例)及びE157E/Q(1例)であった。また、治療下で変異の出現が認められた16例中14例において、試験開始時又はそれ以前からQ148の変異を有していた。
19. 有効成分に関する理化学的知見
| 一般的名称 | ドルテグラビルナトリウム(Dolutegravir Sodium) |
|---|---|
| 化学名 | Monosodium(4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazin-7-olate |
| 分子式 | C20H18F2N3NaO5 |
| 分子量 | 441.36 |
| 性状 | 白色~淡黄白色の粉末。水に溶けにくく、エタノール(99.5)にほとんど溶けない。 |
| 融点 | 1型結晶は約350℃で溶融と同時に分解する。 |
| 化学構造式 | 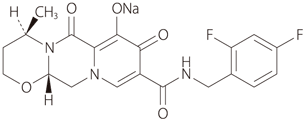 |
| 分配係数 |
2.16±0.01(23℃) |
21. 承認条件
21.1
本剤の使用に当たっては、患者に対して本剤に関して更なる有効性・安全性のデータを引き続き収集中であること等を十分に説明し、インフォームドコンセントを得るよう、医師に要請すること。
21.2
海外において現在実施中又は計画中の臨床試験については、終了後速やかに試験成績及び解析結果を提出すること。
21.3
再審査期間が終了するまでの間、原則として国内の全投与症例を対象とした製造販売後調査を実施し、本剤の使用実態に関する情報(患者背景、有効性・安全性(他剤併用時の有効性・安全性を含む。)及び薬物相互作用のデータ等)を収集して定期的に報告するとともに、調査の結果を再審査申請時に申請書添付資料として提出すること。
22. 包装
30錠[瓶、バラ]
*23. 主要文献
- **Zash R, et al.:International AIDS Conference 2022. Poster PELBB02
- Dickinson L, et al.:Clin Infect Dis.2021;73:e1200-e1207
- Song I, et al.:Antimicrob Agents Chemother.2011;55(7):3517-3521
- Song I, et al.:Eur J Clin Pharmacol.2014;70(10):1173-1179
- Song I, et al.:Antimicrob Agents Chemother.2014;58(11):6696-6700
- 社内資料:薬物相互作用に関する試験(200901)
- Dooley KE, et al.:J Acquir Immune Defic Syndr.2013;62(1):21-27
- Patel P, et al.:J Antimicrob Chemother.2011;66(7):1567-1572
- 社内資料:薬物相互作用に関する試験(201167)
- Moltó J, et al.:Antimicrob Agents Chemother.2016;60(4):2564-2566
- Bollen P, et al.:AIDS.2016;30:1490-1491
- 社内資料:第Ⅰ相試験(ING115381、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.2.1.)
- 社内資料:海外臨床試験(ING111521)
- 社内資料:海外臨床試験(ING111207、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.1.1.)
- 社内資料:海外臨床試験(ING112276)
- 社内資料:分布に関する試験(2011N119355、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.3.1.)
- 社内資料:代謝に関する試験(RD2008/01339、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.6.1.)
- 社内資料:代謝に関する試験(RD2008/00373、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.6.1.)
- Weller S, et al.:Eur J Clin Pharmacol.2014;70(1):29-35
- 社内資料:海外第Ⅰ相試験(ING113097、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.2.2.)
- 社内資料:分布に関する試験(RD2008/00361、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.3.2.)
- 社内資料:分布に関する試験(2011N112380、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.3.2.)
- 社内資料:排泄に関する試験(2010N104937、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.6.2.)
- 社内資料:排泄に関する試験(2013N161621、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.6.2.)
- Raffi F, et al.:Lancet.2013;381(9868):735-743
- 社内資料:海外臨床試験(ING113086)
- 社内資料:海外臨床試験(ING114467)
- Cahn P, et al.:Lancet.2013;382(9893):700-708
- 社内資料:海外臨床試験(ING112574)
**24. 文献請求先及び問い合わせ先
グラクソ・スミスクライン株式会社
東京都港区赤坂1-8-1
ヴィーブヘルスケア・カスタマー・サービス
TEL:0120-066-525(9:00~17:45/土日祝日及び当社休業日を除く)
https://viivhealthcare.com/ja-jp/
26. 製造販売業者等
**26.1 製造販売元
ヴィーブヘルスケア株式会社
東京都港区赤坂1-8-1
**26.2 販売元
グラクソ・スミスクライン株式会社
東京都港区赤坂1-8-1