ビクタルビ配合錠の添付文書
- 商品名:
- ビクタルビ配合錠
- 一般名:
- ビクテグラビル+テノホビル アラフェナミド+エムトリシタビン(BIC/TAF/FTC)
- 略称 :
- BVY

添付文書の読み方
ここで提供している添付文書情報は、2025年2月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
抗ウイルス化学療法剤
ビクテグラビルナトリウム・エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤
- **2025年5月改訂(第6版)
- *2024年9月改訂(第5版)
| 日本標準商品分類番号 | 87625 |
|---|
| 規制区分 | 劇薬 処方箋医薬品注) |
|---|---|
| 貯法 | 室温保存 |
| 有効期間 | 36ヵ月 |
注) 注意―医師等の処方箋により使用すること
| 承認番号 | 23100AMX00302000 |
|---|---|
| 販売開始 | 2019年4月 |
1. 警告
B型慢性肝炎を合併している患者では、本剤の投与中止により、B型慢性肝炎が再燃するおそれがあるので、本剤の投与を中断する場合には十分注意すること。特に非代償性の場合、重症化するおそれがあるので注意すること。
2. 禁忌(次の患者には投与しないこと)
2.1
本剤の成分に対し過敏症の既往歴のある患者
2.2
次の薬剤を投与中の患者:リファンピシン、カルバマゼピン、フェノバルビタール、フェニトイン、ホスフェニトイン、セイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品[10.1 参照]
3. 組成・性状
3.1 組成
| 有効成分・含量 (1錠中) |
ビクテグラビルナトリウム52.5mg(ビクテグラビルとして50mg)、エムトリシタビン200mg及びテノホビル アラフェナミドフマル酸塩28mg(テノホビル アラフェナミドとして25mg) |
|---|---|
| 添加剤 | 結晶セルロース、クロスカルメロースナトリウム、ステアリン酸マグネシウム、黒酸化鉄、三二酸化鉄、マクロゴール4000、ポリビニルアルコール(部分けん化物)、タルク、酸化チタン |
3.2 製剤の性状
| 外形 | 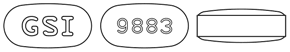 |
|
|---|---|---|
| 大きさ | 長径 | 約15mm |
| 短径 | 約8mm | |
| 重量 | 約721mg | |
| 識別コード | GSI・9883 | |
| 色・剤形 | 紫褐色のフィルムコーティング錠 | |
4. 効能又は効果
HIV-1感染症
5. 効能又は効果に関連する注意
5.1
以下のいずれかのHIV-1感染症患者に使用すること。
5.1.1
抗HIV薬による治療経験がない患者
5.1.2
ウイルス学的失敗の経験がなく、切り替え前3ヵ月間以上においてウイルス学的抑制(HIV-1 RNA量が50copies/mL未満)が得られており、ビクテグラビル、エムトリシタビン又はテノホビルに対する耐性関連変異を持たず、本剤への切り替えが適切であると判断される抗HIV薬既治療患者
**5.1.3
切り替え前6ヵ月間以上においてウイルス学的抑制(HIV-1 RNA量が50copies/mL未満)が得られており、ビクテグラビル又はテノホビルに対する耐性関連変異を持たず、本剤への切り替えが適切であると判断される抗HIV薬既治療患者
**5.2
本剤による治療に当たっては、患者の治療歴及び可能な場合には薬剤耐性検査(遺伝子型解析あるいは表現型解析)を参考にすること。[17.1.5 参照]
6. 用法及び用量
通常、成人には1回1錠(ビクテグラビルとして50mg、エムトリシタビンとして200mg及びテノホビル アラフェナミドとして25mgを含有)を1日1回経口投与する。
7. 用法及び用量に関連する注意
7.1
本剤はビクテグラビルナトリウム、エムトリシタビン及びテノホビル アラフェナミドフマル酸塩の3成分を含有した配合錠である。これらの成分を含む製剤と併用しないこと。また、テノホビル ジソプロキシルフマル酸塩を含む製剤についても併用しないこと。
*7.2
本剤投与後、クレアチニンクリアランスが30mL/分未満に低下した場合は、維持血液透析を行っている患者を除き、投与の中止を考慮すること。なお、透析日に本剤を投与する際は、透析後に1日用量を投与すること。[8.3 参照],[9.2.1 参照],[9.2.2 参照],[11.1.1 参照],[16.6.2 参照]
7.3
本剤はHIV-1感染症に対して1剤で治療を行うものであるため、他の抗HIV薬と併用しないこと。
7.4
エムトリシタビンと類似の薬剤耐性、ウイルス学的特性を有しているラミブジンを含む製剤と併用しないこと。
8. 重要な基本的注意
8.1
本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又はそれに代わる適切な者に次の事項についてよく説明し同意を得た後、使用すること。
8.1.1
本剤はHIV感染症の根治療法薬ではないことから、日和見感染症を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化についてはすべて担当医に報告すること。
8.1.2
本剤の長期投与による影響については現在のところ不明であること。
8.1.3
担当医の指示なしに用量を変更したり、服用を中止したりしないこと。
8.1.4
本剤は併用薬剤と相互作用を起こすことがあるため、服用中のすべての薬剤を担当医に報告すること。また、本剤で治療中に新たに他の薬剤を服用する場合、事前に担当医に相談すること。[10. 参照]
8.2
抗HIV薬の多剤併用療法を行った患者で、免疫再構築炎症反応症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
*8.3
本剤投与前は、クレアチニンクリアランス等の腎機能検査を実施し、クレアチニンクリアランスが30mL/分以上であること又は維持血液透析を行っていることを確認すること。また、本剤投与後も定期的な検査等により、患者の状態を注意深く観察すること。[7.2 参照],[9.2.1 参照],[9.2.2 参照],[10.2 参照],[11.1.1 参照],[16.6.2 参照]
8.4
アジア系人種におけるエムトリシタビンの薬物動態は十分に検討されていないが、少数例の健康成人及びB型慢性肝炎のアジア系人種において、Cmaxの上昇を示唆する成績が得られているので、HBV感染症合併患者を含め、副作用の発現に注意すること。
8.5
エムトリシタビン製剤の試験において皮膚変色が発現し、その発現頻度は有色人種に高いことが示唆されている。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 病的骨折の既往のある患者又はその他の慢性骨疾患を有する患者
十分な観察を行い、異常が認められた場合には、投与を中止する等、適切な処置を行うこと。テノホビル アラフェナミドフマル酸塩を含有する製剤の非臨床試験及び臨床試験において、骨密度の低下と骨代謝の生化学マーカーの上昇が認められ、骨代謝の亢進が示唆された。また、抗HIV薬による治療経験がないHIV-1感染症患者に対し、テノホビル アラフェナミドフマル酸塩を含有する製剤が投与された臨床試験において、骨密度が低下した症例が認められた。
9.1.2 B型肝炎ウイルス感染を合併している患者
本剤の投与を中断する場合には十分注意すること。B型慢性肝炎を合併している患者では、本剤の投与中止により、B型慢性肝炎が再燃するおそれがある。特に非代償性の場合、重症化するおそれがある。
9.1.3 腎機能障害のリスクを有する患者
クレアチニンクリアランス及び血清リンの検査を実施すること。
*9.2 腎機能障害患者
9.2.1 重度の腎機能障害患者
エムトリシタビンの血中濃度が上昇する。維持血液透析を行っていない重度の腎機能障害患者(クレアチニンクリアランスが15mL/分以上30mL/分未満)を対象とした臨床試験は実施していない。[7.2 参照],[8.3 参照],[10.2 参照],[11.1.1 参照],[16.6.2 参照]
9.2.2 末期腎不全患者
エムトリシタビン及びテノホビルの血中濃度が上昇する。ビクテグラビルの血中濃度が低下する。維持血液透析を行っていない末期腎不全患者(クレアチニンクリアランスが15mL/分未満)を対象とした臨床試験は実施していない。[7.2 参照],[8.3 参照],[10.2 参照],[11.1.1 参照],[16.6.2 参照]
9.3 肝機能障害患者
9.3.1 重度の肝機能障害患者
重度の肝機能障害患者を対象とした臨床試験は実施していない。[11.1.2 参照],[16.6.1 参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。他のインテグラーゼ阻害薬であるドルテグラビルでは、海外で進行中の観察研究において、無脳症や二分脊椎などの神経管閉鎖障害が報告されている1)。動物試験(サル)においてテノホビルの胎児への移行が報告されている。
9.6 授乳婦
授乳を避けさせること。動物試験(ラット)でビクテグラビルは乳汁中に分泌され、胎児に移行することが報告されている。テノホビル及びエムトリシタビンはヒト乳汁への移行が報告されている2)。なお、女性のHIV感染症患者は、乳児のHIV感染を避けるため、乳児に母乳を与えないことが望ましい。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能が低下しており、合併症や他の薬剤の併用が多い。
10. 相互作用
ビクテグラビル3),4),5):OCT2及びMATE1を阻害する。CYP3A及びUGT1A1の基質である。
テノホビル及びエムトリシタビン6):糸球体ろ過と能動的な尿細管分泌により腎排泄される。
テノホビル アラフェナミド7):P糖蛋白(P-gp)の基質である。[8.1.4 参照],[16.7.1 参照]
10.1 併用禁忌(併用しないこと)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| リファンピシン (リファジン) [2.2 参照] |
ビクテグラビルの血漿中濃度が低下するため、本剤の効果が減弱し、本剤に対する耐性が発現する可能性があることから、併用しないこと。また、テノホビル アラフェナミドの血漿中濃度も低下する可能性がある。 | リファンピシンのCYP3A、UGT1A1及びP-gpの誘導作用によるため。 |
| カルバマゼピン (テグレトール) フェノバルビタール (フェノバール) フェニトイン (アレビアチン) ホスフェニトイン (ホストイン) セイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品 [2.2 参照] |
ビクテグラビル及びテノホビル アラフェナミドの血漿中濃度が低下するため、本剤の効果が減弱し、本剤に対する耐性が発現する可能性がある。 | これらの薬剤のCYP3A及びP-gpの誘導作用によるため。 |
*10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| ピルシカイニド | ピルシカイニドの血漿中濃度が上昇するため、ピルシカイニドの重大な副作用として報告された心室性頻脈、洞停止及び心室細動等が発現又は増悪する可能性がある。 | ビクテグラビルのOCT2及びMATE1の阻害作用により、ピルシカイニドの排出が阻害される可能性がある。 ピルシカイニドの治療域が狭いため。 |
| リファブチン | ビクテグラビル及びテノホビル アラフェナミドの血漿中濃度が低下するため、本剤の効果が減弱し、本剤に対する耐性が発現する可能性がある。 | これらの薬剤のCYP3A及びP-gpの誘導作用によるため。 |
| アタザナビル | ビクテグラビルの血漿中濃度が上昇する。 | アタザナビルのCYP3A及びUGT1A1の阻害作用によるため。 |
制酸剤
|
ビクテグラビルの血漿中濃度が低下するため、本剤はこれらの製剤の投与2時間以上前の投与が推奨される。 | ビクテグラビルが多価陽イオンと錯体(キレート)を形成し吸収が抑制されるため。 |
| 鉄剤、カルシウム含有製剤(サプリメント等) [16.7.2 参照] |
ビクテグラビルの血漿中濃度が低下するため、これらの製剤を併用する場合は、食後に本剤を投与することが推奨される。 | ビクテグラビルが多価陽イオンと錯体(キレート)を形成し吸収が抑制されるため。 |
| メトホルミン [16.7.2 参照] |
メトホルミンの血漿中濃度が上昇する。注意深く観察し、必要に応じてメトホルミンを減量する等慎重に投与すること。 | ビクテグラビルのOCT2及びMATE1の阻害作用によるため。 |
| アシクロビル バラシクロビル塩酸塩 バルガンシクロビル塩酸塩 |
これらの薬剤、テノホビル又はエムトリシタビンの血漿中濃度が上昇し、これらの薬剤又は本剤による有害事象を増強する可能性がある。 | 排泄経路の競合によるため。 |
| *腎毒性を有する薬剤 [8.3 参照],[9.2.1 参照],[9.2.2 参照],[11.1.1 参照] |
これらの薬剤との併用は避けることが望ましい。 | テノホビル及びエムトリシタビンは主に腎臓での糸球体ろ過と尿細管への能動輸送により排泄されるため。 |
11. 副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1 重大な副作用
11.1.1 腎不全又は重度の腎機能障害(頻度不明)
腎機能不全、腎不全、急性腎障害、近位腎尿細管機能障害、ファンコニー症候群、急性腎尿細管壊死、腎性尿崩症又は腎炎等の重度の腎機能障害があらわれることがあるので、臨床検査値に異常が認められた場合には、投与を中止する等、適切な処置を行うこと。特に腎機能障害の既往がある患者や腎毒性のある薬剤が投与されている患者では注意すること。[7.2 参照],[8.3 参照],[9.2.1 参照],[10.2 参照],[16.6.2 参照]
11.1.2 乳酸アシドーシス及び脂肪沈着による重度の肝腫大(脂肪肝)(頻度不明)
乳酸アシドーシス又は肝細胞毒性が疑われる臨床症状又は検査値異常(アミノトランスフェラーゼの急激な上昇等)が認められた場合には、本剤の投与を一時中止すること。特に肝疾患の危険因子を有する患者においては注意すること。エムトリシタビン又はテノホビルを含む核酸系逆転写酵素阻害薬の単独投与又はこれらの併用療法により、重篤な乳酸アシドーシス及び脂肪沈着による重度の肝腫大(脂肪肝)が多く報告されている。[9.3.1 参照]
11.2 その他の副作用
| 2%以上 | 0.3%以上2%未満 | 頻度不明 | |
|---|---|---|---|
| 心臓障害 | 動悸 | ||
| 神経系障害 | 頭痛、浮動性めまい | 傾眠 | |
| 胃腸障害 | 悪心、下痢 | 便秘、腹部膨満、嘔吐、腹痛、鼓腸、消化不良、腹部不快感、軟便 | |
| 腎及び尿路障害 | 頻尿 | ||
| 皮膚及び皮下組織障害 | 寝汗、脱毛症、そう痒症、発疹 | 血管性浮腫、蕁麻疹 | |
| 筋骨格系及び結合組織障害 | 関節痛 | ||
| 代謝及び栄養障害 | 食欲減退 | 体脂肪の再分布/蓄積 | |
| 血管障害 | ほてり | ||
| 一般・全身障害及び投与部位の状態 | 疲労 | 倦怠感 | |
| 免疫系障害 | 免疫再構築炎症反応症候群 | ||
| 精神障害 | 不眠症、異常な夢、睡眠障害、抑うつ気分、悪夢、リビドー減退 | ||
| 臨床検査 | 腎クレアチニンクリアランス減少 |
13. 過量投与
13.1 処置
ビクテグラビルは血漿蛋白との結合率が高いため、血液透析及び腹膜透析により除去される可能性は低い。エムトリシタビン及びテノホビル アラフェナミドの代謝物であるテノホビルは、血液透析により一部除去される。
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
16. 薬物動態
16.1 血中濃度
16.1.1 健康成人における薬物動態
健康成人に本剤を空腹時に単回経口投与した時の、ビクテグラビル、エムトリシタビン、テノホビル アラフェナミド及びテノホビルの薬物動態パラメータを表16-1に示す8)。
| ビクテグラビル | エムトリシタビン | テノホビル アラフェナミド |
テノホビル | |
|---|---|---|---|---|
| Cmax (μg/mL) |
6.56 (17.9) |
2.68 (39.9) |
0.30 (58.3) |
0.01(30.3) |
| AUCinf (μg・hr/mL) |
115 (21.2) |
11.2 (18.2) |
0.17 (51.6) |
0.33(24.0) |
| t1/2 (hr) |
17.0 (11.8, 23.4) |
18.0 (10.7, 39.1) |
0.35 (0.25, 1.15) |
46.2 (36.1, 106) |
Cmax及びAUCinf:平均値(CV%)
t1/2:中央値(最小値、最大値)
被験者数:25例(テノホビル アラフェナミドのt1/2及びAUCinfは24例)
16.1.2 成人HIV感染症患者における薬物動態
成人HIV感染症患者に本剤を反復経口投与した時の、ビクテグラビル、エムトリシタビン及びテノホビル アラフェナミドの薬物動態パラメータを表16-2に示す(外国人のデータ)9),10),11)。
| ビクテグラビル | エムトリシタビン | テノホビル アラフェナミド | |
|---|---|---|---|
| Cmax (μg/mL) |
6.80 (30.1) |
2.13 (34.7) |
0.277 (62.4) |
| AUCtau (μg・hr/mL) |
94.2 (34.7) |
12.3 (29.2) |
0.229 (63.0) |
| Ctrough (μg/mL) |
2.26 (47.3)b) |
0.096 (37.4)c) |
NA |
| t1/2 (hr) |
16.11 (6.76, 28.32) |
6.82 (2.33, 9.40) |
0.39 (0.17, 1.14) |
Cmax、AUCtau及びCtrough:平均値(CV%)
t1/2:中央値(最小値、最大値)
NA:該当なし
- a)GS-US-380-1489、1490、1844及び1878試験で得られた77例のデータを用いた薬物動態解析(ビクテグラビル及びエムトリシタビンのCtroughを除く)
- b)GS-US-380-1489、1490、1844及び1878試験で得られた75例のデータを用いた薬物動態解析
- c)GS-US-380-1489、1490、1844及び1878試験で得られた74例のデータを用いた薬物動態解析
16.2 吸収
16.2.1 食事の影響
ビクテグラビルを高脂肪食摂取時に投与した場合、空腹時と比べて、AUCinf及びCmaxはそれぞれ約24%及び約13%上昇した。同様に、中脂肪食摂取時のビクテグラビルのAUCinf及びCmaxは、空腹時と比べて上昇した。
食事はエムトリシタビンのAUCinfに影響を及ぼさなかったが、高脂肪食摂取時に投与した場合、Cmaxは空腹時に比べて14%減少した。
テノホビル アラフェナミドを高脂肪食摂取時に投与した場合のAUCinfは、空腹時と比べて約67%上昇した。中脂肪食摂取時のAUCinfは空腹時と比べて約48%上昇したが、Cmaxは影響を受けなかった(外国人のデータ)12)。
16.3 分布
ビクテグラビル13),14):In vitro試験において、ヒト血漿蛋白に対する結合率は99%を超えていた(遊離型の割合は約0.25%)。In vitroヒト血液/血漿中濃度比は0.64であった。
エムトリシタビン15):In vitro試験において、ヒト血漿蛋白に対する結合率は4%未満であり、0.02~200μg/mLの範囲で濃度の影響を受けなかった。
テノホビル アラフェナミド16):臨床試験で採取した検体におけるex vivoでのヒト血漿蛋白に対する結合率は約80%であった。
テノホビル17):In vitro試験において、ヒト血漿蛋白に対する結合率は0.7%未満であり、0.01~25μg/mLの範囲で濃度の影響を受けなかった(外国人のデータ)。
16.4 代謝
ビクテグラビル4),5):ヒトでは、代謝が主な消失経路であり、経口投与されたビクテグラビルの90%超が代謝により消失した。In vitro代謝酵素同定試験において、ビクテグラビルは主にCYP3A及びUGT1A1により代謝されることが示された。
エムトリシタビン6):ヒトでは、エムトリシタビンの代謝は、3’-スルホキシドジアステレオマーを生成(投与量の約9%)するチオール部分の酸化と、2’-O-グルクロニドを生成(投与量の約4%)するグルクロン酸抱合から成ることが示された。
テノホビル アラフェナミド16),18),19),20):ヒトでは、代謝が主な消失経路であり、経口投与されたテノホビル アラフェナミドの80%超が代謝により消失した。In vitro試験において、PBMC(リンパ球及びその他のHIVの標的細胞を含む)及びマクロファージのカテプシンA及び肝細胞のカルボキシルエステラーゼ1によりテノホビルに代謝された。In vivo試験において、細胞内でテノホビル(主要代謝物)に加水分解された後、活性代謝物であるテノホビル二リン酸へとリン酸化された。CYP分子種発現系酵素を用いたin vitro試験において、テノホビル アラフェナミドはCYP3Aでわずかに代謝された(外国人のデータ)。
16.5 排泄
ビクテグラビル21):主に肝代謝により消失し、腎臓からの未変化体のわずかな排泄(投与量の約1%)は主要な経路ではない。14C標識ビクテグラビルを単回経口投与したとき、投与量の約60%が未変化体、デスフルオロ-ヒドロキシ-ビクテグラビル-システイン抱合体及びその他の微量の酸化代謝物として糞中から回収された。尿中回収率は35%であり、主な代謝物はビクテグラビルのグルクロン酸抱合体及びその他の微量の酸化代謝物、並びにそれらの第2相反応の抱合体であった。
エムトリシタビン6):腎クリアランスが推定クレアチニンクリアランスを上回ったことから、糸球体ろ過と尿細管への能動輸送の両方による排泄が示唆された。14C標識エムトリシタビンを投与したときの尿中(約86%)及び糞中(約14%)からの総回収率は100%であった。投与量の13%が3種の推定代謝物として尿中に回収された。
テノホビル アラフェナミド22):テノホビルに代謝された後、排泄される。腎臓からの未変化体のわずかな排泄(投与量の約1%)は主要な経路ではない。テノホビルは腎臓での糸球体ろ過と尿細管への能動輸送の両方により排泄された。健康被験者に14C標識テノホビル アラフェナミドフマル酸塩を単回投与したとき、投与量の47.2%が糞中に、36.2%が尿中に排泄された。その主要代謝物はテノホビルであり、糞中の99%、尿中の86%を占めた。また、投与量の1.4%がテノホビル アラフェナミドとして尿中に排泄された(外国人のデータ)。
*16.6 特定の背景を有する患者
16.6.1 肝機能障害患者における薬物動態
ビクテグラビル23):中等度の肝機能障害を有する被験者にビクテグラビル75mgを単回投与した際のAUCinf及びCmaxは、肝機能正常被験者と比べてそれぞれ41.3%及び36.5%低下した。しかし、中等度肝機能障害被験者のビクテグラビルの血漿蛋白非結合型分率は、肝機能正常被験者と比べて高いため、遊離型ビクテグラビルのAUCinf及びCmaxは両群で同程度であった(最小二乗幾何平均値の比は77~83%)。重度の肝機能障害を有する被験者における薬物動態は検討していない。
エムトリシタビン:肝機能障害を有する被験者における薬物動態は検討していない。
テノホビル アラフェナミド24),25):肝機能正常被験者と比べて、軽度又は中等度の肝機能障害を有する被験者にテノホビル アラフェナミド25mgを単回投与した際のテノホビル アラフェナミドのAUCinf及びCmaxは、軽度の肝機能障害を有する被験者ではそれぞれ7.5%及び11.0%低下し、中等度の肝機能障害を有する被験者ではそれぞれ12.7%及び18.7%上昇した。肝機能正常被験者と比べて、軽度又は中等度の肝機能障害を有する被験者にテノホビル アラフェナミド25mgを単回投与した際のテノホビルのAUCinf及びCmaxは、軽度の肝機能障害を有する被験者ではそれぞれ10.8%及び3.0%低下し、中等度の肝機能障害を有する被験者ではそれぞれ2.8%及び12.4%低下した。肝機能正常被験者と比べて、重度の肝機能障害を有する被験者にテノホビル アラフェナミド25mgを単回投与した際のテノホビル アラフェナミドの結合型及び非結合型の総AUCinf及びCmaxはそれぞれ46.0%及び54.9%低下し、テノホビルのAUCinf及びCmaxはそれぞれ36.9%及び10.1%低下した。蛋白結合率(重度の肝機能障害を有する被験者で37.8%、肝機能正常被験者で20.4%)で補正したとき、重度の肝機能障害を有する被験者における遊離型(非結合型)テノホビル アラフェナミドのAUCinf及びCmaxは、肝機能正常被験者と比べてそれぞれ5.6%及び17.8%低下した(外国人のデータ)。[9.3.1 参照]
16.6.2 腎機能障害患者における薬物動態
ビクテグラビル26),73):重度の腎機能障害(クレアチニンクリアランス:15mL/分以上30mL/分未満)を有する被験者にビクテグラビル75mgを単回投与した際の血漿中ビクテグラビルの結合型及び非結合型の総AUCinf及びCmaxは、腎機能正常被験者と比べてそれぞれ27%及び20%低下した。重度の腎機能障害を有する被験者のビクテグラビルの血漿蛋白非結合型分率は、腎機能正常被験者と比べて高いため、遊離型ビクテグラビルのAUCinf及びCmaxは両群間で同程度であった。維持血液透析を行っている末期腎不全(クレアチニンクリアランス:15mL/分未満)を有するHIV-1感染症患者に本剤を1日1回反復投与した際の血漿中ビクテグラビルの結合型及び非結合型の総Ctroughは、腎機能正常HIV-1感染症患者と比べて65%低かった74)。
エムトリシタビン27),73):重度の腎機能障害(クレアチニンクリアランス:15mL/分以上30mL/分未満)を有する被験者にエムトリシタビン200mgを単回投与した際のAUC及びCmaxは、クレアチニンクリアランスが80mL/分超の被験者と比べて、それぞれ約200%及び約30%上昇した。維持血液透析を行っている末期腎不全(クレアチニンクリアランス15mL/分未満)を有するHIV-1感染症患者にエルビテグラビル・コビシスタット・エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤(150・150・200・10mg)を1日1回反復投与した際のAUCtau及びCmaxは、腎機能正常HIV-1感染症患者と比べてそれぞれ438%及び137%上昇した75)。
テノホビル アラフェナミド28),29),73):腎機能正常被験者と比べて、重度の腎機能障害(クレアチニンクリアランス:15mL/分以上30mL/分未満)を有する被験者にテノホビル アラフェナミド25mgを単回投与した際のテノホビル アラフェナミドのAUCinf及びCmaxは、それぞれ1.9倍及び1.8倍上昇し、テノホビルのAUCinf及びCmaxは、それぞれ5.7倍及び2.8倍上昇した。腎機能正常HIV-1感染症患者と比べて、維持血液透析を行っている末期腎不全(クレアチニンクリアランス:15mL/分未満)を有するHIV-1感染症患者にエルビテグラビル・コビシスタット・エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤(150・150・200・10mg)を1日1回反復投与した際のテノホビルのAUCtau及びCmaxは、それぞれ27.0倍及び2.4倍75)、テノホビル アラフェナミドのAUClast及びCmaxは、それぞれ1.0倍及び1.1倍であった75)(外国人のデータ)。[7.2 参照],[8.3 参照],[9.2.1 参照],[9.2.2 参照],[11.1.1 参照]
16.7 薬物相互作用
16.7.1 非臨床における薬物相互作用試験
ビクテグラビル3),4),5):ビクテグラビルはP-gp及びBCRPの基質であり、OCT2及びMATE1に対する阻害作用を示した。
エムトリシタビン30):In vitroではOAT3の基質である。
テノホビル アラフェナミド7):テノホビル アラフェナミドは、P-gp及びBCRPの基質である。[10. 参照]
16.7.2 臨床における薬物相互作用試験
薬物相互作用試験の結果を、表16-3から表16-7に示す(外国人のデータ)。[10.2 参照]
| 併用薬 | 併用薬の用量 | ビクテグラビル | 例数 | 他剤併用時/非併用時のビクテグラビルの 薬物動態パラメータ比(90%信頼区間) |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| アタザナビル(食後)31) | 300mg+ コビシスタット 150mg 1日1回 |
75mg 単回 |
15 | 1.31 (1.23, 1.40) |
4.06 (3.76, 4.37) |
NA |
| アタザナビル(食後)31) | 400mg 1日1回 |
75mg 単回 |
15 | 1.28 (1.23, 1.33) |
4.15 (3.81, 4.51) |
NA |
| ダルナビル(食後)31) | 800mg+ コビシスタット 150mg 1日1回 |
75mg 1日1回 |
13 | 1.52 (1.40, 1.64) |
1.74 (1.62, 1.87) |
2.11 (1.95, 2.29) |
| レジパスビル・ソホスブビル(食後)32) | 90/400mg 1日1回 |
75mg 1日1回b |
30 | 0.98 (0.94, 1.03) |
1.00 (0.97, 1.03) |
1.04 (0.99, 1.09) |
| リファブチン(空腹時)31) | 300mg 1日1回 |
75mg 1日1回 |
13 | 0.80 (0.67, 0.97) |
0.62 (0.53, 0.72) |
0.44 (0.37, 0.52) |
| リファンピシン(食後)31) | 600mg 1日1回 |
75mg 単回 |
15 | 0.72 (0.67, 0.78) |
0.25 (0.22, 0.27) |
NA |
| ソホスブビル・ベルパタスビル /voxilaprevir(食後)33) |
400mg/100mg/ 100mg+100mg voxilaprevirc 1日1回 |
50mg 1日1回b |
30 | 0.98 (0.94, 1.01) |
1.07 (1.03, 1.10) |
1.10 (1.05, 1.17) |
| ボリコナゾール(空腹時)31) | 300mg 1日2回 |
75mg 単回 |
15 | 1.09 (0.96, 1.23) |
1.61 (1.41, 1.84) |
NA |
| 高用量制酸剤 (本剤と同時投与/空腹時)34) |
20mLd 単回(経口) |
50mg 単回b |
14 | 0.20 (0.16, 0.24) |
0.21 (0.18, 0.26) |
NA |
| 高用量制酸剤 (本剤投与の2時間後/空腹時)34) |
20mLd 単回(経口) |
50mg 単回b |
13 | 0.93 (0.88, 1.00) |
0.87 (0.81, 0.93) |
NA |
| 高用量制酸剤 (本剤投与の2時間前/空腹時)34) |
20mLd 単回(経口) |
50mg 単回b |
13 | 0.42 (0.33, 0.52) |
0.48 (0.38, 0.59) |
NA |
| 高用量制酸剤 (本剤と同時投与/食後e)34) |
20mLd 単回(経口) |
50mg 単回b |
14 | 0.51 (0.43, 0.62) |
0.53 (0.44, 0.64) |
NA |
| 炭酸カルシウム (本剤と同時投与/空腹時)34) |
1200mg 単回 |
50mg 単回b |
14 | 0.58 (0.51, 0.67) |
0.67 (0.57, 0.78) |
NA |
| 炭酸カルシウム (本剤と同時投与/食後e)34) |
1200mg 単回 |
50mg 単回b |
14 | 0.90 (0.78, 1.03) |
1.03 (0.89, 1.20) |
NA |
| フマル酸第一鉄 (本剤と同時投与/空腹時)34) |
324mg 単回 |
50mg 単回b |
14 | 0.29 (0.26, 0.33) |
0.37 (0.33, 0.42) |
NA |
| フマル酸第一鉄 (本剤と同時投与/食後e)34) |
324mg 単回 |
50mg 単回b |
14 | 0.75 (0.65, 0.87) |
0.84 (0.74, 0.95) |
NA |
NA=該当なし
- a. いずれの相互作用試験も健康被験者を対象として実施した。
- b. ビクテグラビルナトリウム・エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤を用いて試験を実施した。
- c. HCV感染症患者で予測されるvoxilaprevirの曝露量を達成するため、voxilaprevir 100mgを追加増量して試験を実施した。
- d. 高用量制酸剤は、1mL中に水酸化アルミニウム80mg、水酸化マグネシウム80mg及びシメチコン8mgを含有する。
- e. 比較対照の投与は空腹時に行った。
| 併用薬 | 併用薬の用量 | エムトリシタビン | 例数 | 他剤併用時/非併用時のエムトリシタビンの 薬物動態パラメータ比(90%信頼区間) |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| レジパスビル・ソホスブビル(食後)32) | 90mg/400mg 1日1回 |
200mg 1日1回b |
30 | 0.99 (0.94, 1.05) |
0.99 (0.95, 1.02) |
1.03 (0.99, 1.07) |
| ソホスブビル・ベルパタスビル /voxilaprevir(食後)33) |
400mg/100mg/ 100mg+100mg voxilaprevirc 1日1回 |
200mg 1日1回b |
30 | 0.89 (0.83, 0.94) |
0.95 (0.93, 0.97) |
1.10 (1.05, 1.16) |
- a. いずれの相互作用試験も健康被験者を対象として実施した。
- b. ビクテグラビルナトリウム・エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤を用いて試験を実施した。
- c. HCV感染症患者で予測されるvoxilaprevirの曝露量を達成するため、voxilaprevir 100mgを追加増量して試験を実施した。
| 併用薬 | 併用薬の用量 | テノホビル アラフェナミド | 例数 | 他剤併用時/非併用時のテノホビル アラフェナミドの 薬物動態パラメータ比(90%信頼区間) |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| カルバマゼピン35) | 300mg 1日2回 |
25mg 単回b |
22 | 0.43 (0.36, 0.51) |
0.46 (0.40, 0.54) |
NA |
| レジパスビル・ソホスブビル(食後)32) | 90mg/400mg 1日1回 |
25mg 1日1回c |
30 | 1.17 (1.00, 1.38) |
1.27 (1.19, 1.34) |
NA |
| ソホスブビル・ベルパタスビル /voxilaprevir(食後)33) |
400mg/100mg/ 100mg+100mg voxilaprevird 1日1回 |
25mg 1日1回c |
30 | 1.28 (1.09, 1.51) |
1.57 (1.44, 1.71) |
NA |
NA = 該当なし
- a. いずれの相互作用試験も健康被験者を対象として実施した。
- b. エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤を用いて試験を実施した。
- c. ビクテグラビルナトリウム・エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤を用いて試験を実施した。
- d. HCV感染症患者で予測されるvoxilaprevirの曝露量を達成するため、voxilaprevir 100mgを追加増量して試験を実施した。
| 併用薬 | 併用薬の用量 | テノホビル アラフェナミド | 例数 | 他剤併用時/非併用時のテノホビルの 薬物動態パラメータ比(90%信頼区間) |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| カルバマゼピン35) | 300mg 1日2回 |
25mg 単回b |
22 | 0.70 (0.65, 0.74) |
0.77 (0.74, 0.81) |
NA |
| レジパスビル・ソホスブビル(食後)32) | 90mg/400mg 1日1回 |
25mg 1日1回c |
30 | 1.43 (1.37, 1.50) |
1.67 (1.60, 1.74) |
1.81 (1.73, 1.90) |
| ソホスブビル・ベルパタスビル /voxilaprevir(食後)33) |
400mg/100mg/ 100mg+100mg voxilaprevird 1日1回 |
25mg 1日1回c |
30 | 1.51 (1.45,1.58) |
1.67 (1.62,1.73) |
1.74 (1.68, 1.80) |
NA = 該当なし
- a. いずれの相互作用試験も健康被験者を対象として実施した。
- b. エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤を用いて試験を実施した。
- c. ビクテグラビルナトリウム・エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤を用いて試験を実施した。
- d. HCV感染症患者で予測されるvoxilaprevirの曝露量を達成するため、voxilaprevir 100mgを追加増量して試験を実施した。
| 併用薬 | 併用薬の用量 | ビクテグラビル | テノホビル アラフェナミド | 例数 | 併用薬の薬物動態パラメータ比 (90%信頼区間CI) |
||
|---|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | |||||
| レジパスビル32) | 90mg/400mg 1日1回 |
75mg 1日1回c |
25mg 1日1回c |
30 | 0.85 (0.81, 0.90) |
0.87 (0.83, 0.92) |
0.90 (0.84, 0.96) |
| ソホスブビル32) | 1.11 (1.00, 1.24) |
1.07 (1.01, 1.13) |
NA | ||||
| GS-331007 b,32) | 1.10 (1.07, 1.13) |
1.11 (1.08, 1.14) |
1.02 (0.99, 1.06) |
||||
| メトホルミン36) | 500mg 1日2回 |
50mg 1日1回c |
25mg 1日1回c |
30 | 1.28 (1.21, 1.36) |
1.39 (1.31, 1.48) |
1.36 (1.21, 1.53) |
| ミダゾラム37) | 2mg 単回 |
50mg 1日1回c |
25mg 1日1回c |
14 | 1.03 (0.87, 1.23) |
1.15 (1.00, 1.31) |
NA |
| Norelgestromin38) | norgestimate 0.180mg/0.215mg/0.250mg 1日1回 エチニルエストラジオール 0.025mg 1日1回 |
75mg 1日1回 |
- | 15 | 1.23 (1.14, 1.32) |
1.08 (1.05, 1.10) |
1.10 (1.05, 1.15) |
| ノルゲストレル38) | 1.15 (1.10, 1.21) |
1.13 (1.07, 1.19) |
1.14 (1.06, 1.22) |
||||
| エチニルエストラジオール38) | 1.15 (1.03, 1.27) |
1.04 (0.99, 1.10) |
1.05 (0.95, 1.14) |
||||
| Norelgestromin38) | norgestimate 0.180mg/0.215mg/0.250mg 1日1回 エチニルエストラジオール 0.025mg 1日1回 |
- | 25mg 1日1回d |
14 | 1.17 (1.07,1.26) |
1.12 (1.07,1.17) |
1.16 (1.08, 1.24) |
| ノルゲストレル38) | 1.10 (1.02, 1.18) |
1.09 (1.01, 1.18) |
1.11 (1.03, 1.20) |
||||
| エチニルエストラジオール38) | 1.22 (1.15, 1.29) |
1.11 (1.07, 1.16) |
1.02 (0.92, 1.12) |
||||
| セルトラリン39) | 50mg 単回 |
- | 10mg 1日1回e |
19 | 1.14 (0.94, 1.38) |
0.93 (0.77, 1.13) |
NA |
| ソホスブビル33) | 400mg/100mg/100mg+100mgf 1日1回 |
50mg 1日1回c |
25mg 1日1回c |
30 | 1.14 (1.04, 1.25) |
1.09 (1.02, 1.15) |
NA |
| GS-331007b,33) | 1.03 (0.99, 1.06) |
1.03 (1.00, 1.06) |
1.01 (0.98, 1.05) |
||||
| ベルパタスビル33) | 0.96 (0.91, 1.01) |
0.96 (0.90, 1.02) |
0.94 (0.88, 1.01) |
||||
| Voxilaprevir33) | 0.90 (0.76, 1.06) |
0.91 (0.80, 1.03) |
0.97 (0.88, 1.06) |
||||
NA = 該当なし
- a. いずれの相互作用試験も健康被験者を対象として実施した。
- b. 循環血液中のソホスブビルの主要ヌクレオシド代謝物
- c. ビクテグラビルナトリウム・エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤を用いて試験を実施した。
- d. エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤を用いて試験を実施した。
- e. エルビテグラビル・コビシスタット・エムトリシタビン・テノホビル アラフェナミドフマル酸塩配合剤を用いて試験を実施した。
- f. HCV感染症患者で予測されるvoxilaprevirの曝露量を達成するため、voxilaprevir 100mgを追加増量して試験を実施した。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相臨床試験
抗HIV薬による治療経験がないHIV-1感染症患者を対象とし、本剤の有効性及び安全性を検討することを目的として、ドルテグラビル(DTG)・アバカビル(ABC)・ラミブジン(3TC)レジメンを対照とした無作為化二重盲検並行群間比較試験[GS-US-380-1489試験(1489試験)]を実施した(外国人のデータ)40)。治験薬投与48週時の結果は表17-1のとおりであり、対照に対する本剤の非劣性が検証された(非劣性マージン:-12%)。
| 1489試験 | ||
|---|---|---|
| 本剤群 | DTG/ABC/3TC群 | |
| HIV-1 RNA量が50copies/mL 未満の被験者の割合a) |
92.4% (290/314例) |
93.0% (293/315例) |
| 群間差[95.002%信頼区間]a, b) | -0.6[-4.8, 3.6]% | |
| ウイルス学的失敗例の割合 a,c) | 1.0% (3/314例) |
2.5% (8/315例) |
- a)欠測値はFDA snapshot algorithmに従い取り扱われた。
- b)スクリーニング時点でのHIV-1 RNA量(100,000copies/mL以下、100,000copies/mL超)及び地域(米国、米国以外)を層別因子として調整。
- c)以下①~③のいずれかに該当した被験者。
①投与48週時のHIV-1 RNA量50copies/mL以上、②有効性の欠如による中止例、③有効性の欠如以外の理由による中止例のうち、最終検査時にHIV-1 RNA量50copies/mL以上
本剤を投与された314例中82例(26.1%)に臨床検査値異常を含む副作用が認められた。主な副作用は、下痢19例(6.1%)、悪心17例(5.4%)、頭痛16例(5.1%)等であった(承認時)。
17.1.2 国際共同第III相臨床試験
抗HIV薬による治療経験がないHIV-1感染症患者を対象とし、本剤の有効性及び安全性を検討することを目的として、DTG+エムトリシタビン(FTC)・テノホビル アラフェナミド(TAF)レジメンを対照とした無作為化二重盲検並行群間比較試験[GS-US-380-1490試験(1490試験)]を実施した(外国人のデータ)41)。治験薬投与48週時の結果は表17-2のとおりであり、対照に対する本剤の非劣性が検証された(非劣性マージン:-12%)。
| 1490試験 | ||
|---|---|---|
| 本剤群 | DTG+FTC/TAF群 | |
| HIV-1 RNA量が50copies/mL 未満の被験者の割合a) |
89.4% (286/320例) |
92.9% (302/325例) |
| 群間差[95.002%信頼区間]a,b) | -3.5[-7.9, 1.0]% | |
| ウイルス学的失敗例の割合a,c) | 4.4%(14/320例) | 1.2%(4/325例) |
- a)欠測値はFDA snapshot algorithmに従い取り扱われた。
- b)スクリーニング時点でのHIV-1 RNA量(100,000copies/mL以下、100,000copies/mL超)及び地域(米国、米国以外)を層別因子として調整。
- c)以下①~③のいずれかに該当した被験者。
①投与48週時のHIV-1 RNA量50copies/mL以上、②有効性の欠如による中止例、③有効性の欠如以外の理由による中止例のうち、最終検査時にHIV-1 RNA量50copies/mL以上
本剤を投与された320例中57例(17.8%)に臨床検査値異常を含む副作用が認められた。主な副作用は、頭痛13例(4.1%)、下痢10例(3.1%)、悪心9例(2.8%)等であった(承認時)。
17.1.3 国際共同第III相臨床試験
ドルテグラビル・アバカビル・ラミブジン(DTG/ABC/3TC)レジメンにより3カ月以上持続してウイルス学的抑制(HIV-1 RNA量50copies/mL未満)が得られているHIV-1感染症患者を対象とし、本剤に切替えた場合の有効性及び安全性を検討することを目的として、スクリーニング時に施行されていたレジメン継続投与を対照とした無作為化二重盲検並行群間比較試験[GS-US-380-1844試験(1844試験)]を実施した(外国人のデータ)42)。治験薬投与48週時の結果は表17-3のとおりであり、対照に対する本剤の非劣性が検証された(非劣性マージン:4%)。
| 1844試験 | ||
|---|---|---|
| 本剤群 | DTG/ABC/3TC継続群 | |
| ウイルス学的失敗例の割合 a,b) | 1.1%(3/282例) | 0.4%(1/281例) |
| 群間差[95.002%信頼区間]a) | 0.7[-1.0, 2.8]% | |
| HIV-1 RNA量が50copies/mL 未満の被験者の割合a) |
93.6%(264/282例) | 95.0%(267/281例) |
- a)欠測値はFDA snapshot algorithmに従い取り扱われた。
- b)以下①~③のいずれかに該当した被験者。
①投与48週時のHIV-1 RNA量50copies/mL以上、②有効性の欠如による中止例、③有効性の欠如以外の理由による中止例のうち、最終検査時にHIV-1 RNA量50copies/mL以上
本剤を投与された282例中23例(8.2%)に臨床検査値異常を含む副作用が認められた。主な副作用は、頭痛7例(2.5%)等であった(承認時)。
**17.1.4 国際共同第III相臨床試験
リトナビル(RTV)又はコビシスタット(COBI)+プロテアーゼ阻害薬[PI:アタザナビル(ATV)又はダルナビル(DRV)]+核酸系逆転写酵素阻害薬[NRTI:エムトリシタビン・テノホビル ジソプロキシルフマル酸塩(TDF)又はABC/3TC]の併用レジメンにより、6カ月以上持続してウイルス学的抑制(HIV-1 RNA量50copies/mL未満)が得られているHIV-1感染症患者を対象とし、本剤に切替えた場合の有効性及び安全性を検討することを目的として、スクリーニング時に施行されていたレジメン継続投与を対照とした無作為化二重盲検並行群間比較試験[GS-US-380-1878試験(1878試験)]を実施した(外国人のデータ)43)。治験薬投与48週時の結果は表17-4のとおりであり、対照に対する本剤の非劣性が検証された(非劣性マージン:4%)。
| 1878試験 | ||
|---|---|---|
| 本剤群 | PI+NRTI継続群 | |
| ウイルス学的失敗例の割合a,b) | 1.7%(5/290例) | 1.7%(5/287例) |
| 群間差[95.002%信頼区間]a) | -0.0[-2.5, 2.5]% | |
| HIV-1 RNA量が50copies/mL 未満の被験者の割合a) |
92.1%(267/290例) | 88.9%(255/287例) |
- a)欠測値はFDA snapshot algorithmに従い取り扱われた。
- b)以下①~③のいずれかに該当した被験者。
①投与48週時のHIV-1 RNA量50copies/mL以上、②有効性の欠如による中止例、③有効性の欠如以外の理由による中止例のうち、最終検査時にHIV-1 RNA量50copies/mL以上
本剤を投与された290例中54例(18.6%)に臨床検査値異常を含む副作用が認められた。主な副作用は、頭痛14例(4.8%)、鼓腸及び悪心各2例(2.4%)等であった(承認時)。
**17.1.5 国際共同第III相臨床試験
DTG+FTC/TAF又はDTG+FTC/TDFの併用レジメンにより、スクリーニング時点で6カ月以上(NRTI変異が確認された又は疑われる場合)又は3カ月以上(NRTI変異が確認されていない又は疑われない場合)継続してウイルス学的抑制(HIV-1 RNA量50copies/mL未満)が得られているHIV-1感染症患者を対象とし、本剤に切替えた場合の有効性及び安全性を検討することを目的として、DTG+FTC/TAFレジメンを対照とした無作為化二重盲検並行群間比較試験[GS-US-380-4030試験(4030試験)]を実施した(外国人のデータ)76)。治験薬投与48週時の結果は表17-5のとおりであり、対照に対する本剤の非劣性が検証された(非劣性マージン:4%)。また、本剤を投与された284例中47例はベースライン時点でFTCに対する耐性関連変異であるM184V/I変異が認められ、M184V/I変異が認められた被験者の89%(47例中42例)では48週時点でウイルス抑制が維持された。11%(47例中5例)では治験薬の投与中止により48週時点のウイルス学的データが得られなかったが、最終検査時のHIV-1 RNA量は50copies/mL未満であった。
| 4030試験 | ||
|---|---|---|
| 本剤群 | DTG+F/TAF群 | |
| ウイルス学的失敗例の割合a),b) | 0.4%(1/284例) | 1.1%(3/281例) |
| 群間差[95.001%信頼区間]a) | -0.7[-2.8, 1.0]% | |
| HIV-1 RNA量が50copies/mL 未満の被験者の割合a) |
93.3%(265/284例) | 91.1%(256/281例) |
- a)欠測値はFDA snapshot algorithmに従い取り扱われた。
- b)以下①~③のいずれかに該当した被験者。
①投与48週時のHIV-1 RNA量50copies/mL以上、②有効性の欠如による中止例、③有効性の欠如以外の理由による中止例のうち、最終検査時にHIV-1 RNA量50copies/mL以上
本剤を投与された284例中46例(16.2%)に臨床検査値異常を含む副作用が認められた。主な副作用は、異常な夢及び体重増加各5例(1.8%)等であった(48週時点)。[5.2 参照]
17.3 その他
17.3.1 心電図に対する影響
ビクテグラビル44):健康被験者48例を対象として心電図に対する影響を評価し、推奨治療用量の1.5倍及び6倍の用量でビクテグラビルを投与したところ、QT/QTc間隔に影響を与えず、PR間隔を延長させなかった。
エムトリシタビン:エムトリシタビンがQT/QTc間隔に与える影響は不明である。
テノホビル アラフェナミド45):健康被験者48例を対象として心電図に対する影響を評価し、推奨治療用量及びその5倍の用量でテノホビル アラフェナミドを投与したところ、QT/QTc間隔に影響を与えず、PR間隔を延長させなかった(外国人のデータ)。
18. 薬効薬理
18.1 作用機序
ビクテグラビル46),47),48),49):ビクテグラビルはインテグラーゼ阻害剤であり、インテグラーゼの活性部位と結合し、HIVの複製サイクルに必要なレトロウイルスDNAの組込み過程におけるDNA鎖のトランスファーを阻害する。
エムトリシタビン50),51),52):エムトリシタビンは、核酸系逆転写酵素阻害剤(NRTI)であり、細胞内酵素によりリン酸化されエムトリシタビン5’-三リン酸となる。エムトリシタビン5’-三リン酸は、HIV逆転写酵素によるウイルスDNAへの取込みを介してHIV複製を阻害し、DNA鎖の伸長を停止させる。
テノホビル アラフェナミド20),21),22),53),54),55),56):テノホビル アラフェナミドは、NRTIであり、テノホビルのプロドラッグである。テノホビル アラフェナミドは、PBMC及びマクロファージ中のカテプシンAにより加水分解を受けて細胞内にテノホビルを送達する。その後、細胞内でリン酸化を受け、テノホビル二リン酸となり、HIV逆転写酵素によるウイルスDNAへの取込みを介してHIV複製を阻害し、DNA鎖の伸長を停止させる。
18.2 抗ウイルス活性
ビクテグラビル、エムトリシタビン及びテノホビル アラフェナミドを細胞培養系で評価した結果、相乗的な抗ウイルス活性が認められた。
ビクテグラビル46),47),57),58):ヒトTリンパ芽球様細胞株、PBMC、単球/マクロファージ初代培養細胞及びCD4陽性Tリンパ球を用いて、HIV-1の実験室株及び臨床分離株に対するビクテグラビルの抗ウイルス活性を評価した。ビクテグラビルの50%効果濃度(EC50値)は0.05nmol/L未満~6.6nmol/Lの範囲であった。野生型HIV-1に対するビクテグラビルのタンパク質補正EC95値は361nmol/L(0.162μg/mL)であった。HIV-1グループM、N及びO(サブタイプA~Gを含む)に対するビクテグラビルのEC50値は0.05nmol/L未満~1.71nmol/Lの範囲であった。HIV-2に対するEC50値は1.1nmol/Lであった。
エムトリシタビン59),60),61):ヒトTリンパ芽球様細胞株、MAGI-CCR5細胞株及びPBMCを用いて、HIV-1の実験室株及び臨床分離株に対するエムトリシタビンの抗ウイルス活性を評価した。エムトリシタビンのEC50値は0.0013~0.64μmol/Lの範囲であった。HIV-1サブタイプA~Gに対するエムトリシタビンのEC50値は0.007~0.075μmol/Lの範囲であった。HIV-2に対するEC50値は0.007~1.5μmol/Lの範囲であった。
テノホビル アラフェナミド62):ヒトTリンパ芽球様細胞株、PBMC、単球/マクロファージ初代培養細胞及びCD4陽性Tリンパ球を用いて、HIV-1の実験室株及び臨床分離株に対するテノホビル アラフェナミドの抗ウイルス活性を評価した。テノホビル アラフェナミドのEC50値は2.0~14.7nmol/Lの範囲であった。HIV-1グループM、N及びO(サブタイプA~Gを含む)に対するテノホビル アラフェナミドのEC50値は0.10~12.0nmol/Lの範囲であった。HIV-2に対するEC50値は0.91~2.63nmol/Lの範囲であった。
18.3 薬剤耐性
18.3.1 In vitro試験
ビクテグラビル63),64):ビクテグラビルに対する感受性が低下したHIV-1株では、M50I、R263K及びS153F変異が認められた。
エムトリシタビン65):エムトリシタビンに対する感受性の低下は、HIV-1逆転写酵素のM184V/I変異と関連が認められた。
テノホビル アラフェナミド66):テノホビル アラフェナミドに対する感受性が低下したHIV-1分離株では、K65R変異が発現しており、K70E 変異も一過性に認められた。
18.3.2 臨床試験
抗HIV薬による治療経験のないHIV-1感染症患者:1489試験及び1490試験の併合解析では、ウイルス学的失敗と判定された時点、投与48週後又は早期に試験中止となった時点のHIV-1 RNA量が200copies/mL以上であった8例の遺伝子型及び表現型解析において、新たな耐性変異の発現は認められなかった。
抗HIV薬による治療経験があり、ウイルス学的に抑制されているHIV-1感染症患者:1878試験では、ウイルス学的失敗と判定された時点、投与48週後又は早期に試験中止となった時点のHIV-1 RNA量が200copies/mL以上であった1例の遺伝子型及び表現型解析において、新たな耐性変異の発現は認められなかった(外国人のデータ)。
18.4 交差耐性
ビクテグラビル67),68):G140A/C/S及びQ148H/R/Kの両変異を有するHIV-1株(14株)は、ビクテグラビルに対する感受性が2.5倍を超えて低下した。これら14株のうち9株では、さらにL74M、T97A又はE138A/Kの変異が認められた。また、G118R及びT97A+G118Rの部位特異的変異を導入したHIV-1株では、ビクテグラビルに対する感受性がそれぞれ3.4倍及び2.8倍に低下した。INSTI耐性関連変異(E92Q、T97A、Y143C/R、Q148R、N155H等)を有するHIV-1株では、ビクテグラビルに対する感受性が2倍未満に低下した。
エムトリシタビン69),70):M184V/I変異を有するエムトリシタビン耐性株はラミブジンに対する交差耐性を示した。また、アバカビル、ジダノシン及びテノホビルの投与により出現したK65R変異を有するHIV-1株は、エムトリシタビンに対する感受性の低下を示した。
テノホビル アラフェナミド71),72):K65R及びK70E変異を持つHIV-1株は、アバカビル、ジダノシン、ラミブジン、エムトリシタビン及びテノホビルに対する感受性の低下を示したが、ジドブジンに対する感受性を維持した。T69S二重挿入変異又はK65Rを含むQ151M複合変異を持ち、NRTIに多剤耐性を示すHIV-1は、テノホビル アラフェナミドに対する感受性の低下を示した。
19. 有効成分に関する理化学的知見
| 一般的名称 | ビクテグラビルナトリウム Bictegravir sodium(JAN) |
|---|---|
| 化学名 | Monosodium(2R,5S,13aR)-7,9-dioxo-10-{[(2,4,6-trifluorophenyl)methyl]carbamoyl}-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepin-8-olate |
| 分子式 | C21H17F3N3NaO5 |
| 分子量 | 471.36 |
| 性状 | 微黄白色~黄色の固体 |
| 化学構造式 | 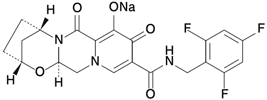 |
| 融点 | 約330℃(分解) |
| 分配係数 | Log P=1.45(1-オクタノール/pH 6.0の緩衝液) |
| 溶解性 | 酢酸に溶けやすく、N-メチルピロリジノンにやや溶けにくく、ジメチルスルホキシドに溶けにくく、水(pH 8.8)又はメタノールに極めて溶けにくく、水(pH 1.8-7.8)、エタノール又はジクロロメタンにほとんど溶けない。 |
| 一般的名称 | エムトリシタビン Emtricitabine(JAN) |
|---|---|
| 化学名 | 4-Amino-5-fluoro-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]pyrimidin-2(1H)-one |
| 分子式 | C8H10FN3O3S |
| 分子量 | 247.25 |
| 性状 | 白色~帯黄白色の粉末 |
| 化学構造式 |  |
| 融点 | 約155℃ |
| 分配係数 | Log P=-0.43(オクタノール/水) |
| 溶解性 | 水、メタノールに溶けやすく、アセトニトリルに溶けにくく、酢酸イソプロピルに極めて溶けにくい。 |
| 一般的名称 | テノホビル アラフェナミドフマル酸塩 Tenofovir alafenamide fumarate(JAN) |
|---|---|
| 化学名 | 1-Methylethyl N-[(S)-{[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl}phenoxyphosphinoyl]-L-alaninate hemifumarate |
| 分子式 | (C21H29N6O5P)2・C4H4O4 |
| 分子量 | 1069.00 |
| 性状 | 白色~灰白色又は白色~くすんだ黄赤色の粉末 |
| 化学構造式 | 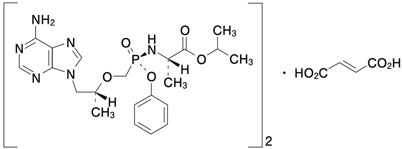 |
| 融点 | 約132℃ |
| 分配係数 | Log P=1.6(1-オクタノール/pH 7のリン酸塩緩衝液) |
| 溶解性 | メタノールに溶けやすく、エタノール(99.5)にやや溶けやすく、水又は2-プロパノールにやや溶けにくく、アセトニトリル又はアセトンに溶けにくく、トルエンに極めて溶けにくい。 |
20. 取扱い上の注意
開封後は湿気を避けて保存すること。
21. 承認条件
21.1
医薬品リスク管理計画を策定の上、適切に実施すること。
21.2
本剤の使用に当たっては、患者に対して本剤に関して更なる有効性・安全性のデータを引き続き収集中であること等を十分に説明し、インフォームドコンセントを得るよう、医師に要請すること。
21.3
海外において現在実施中又は計画中の臨床試験については、終了後速やかに試験成績及び解析結果を提出すること。
21.4
再審査期間が終了するまでの間、原則として国内の全投与症例を対象とした製造販売後調査を実施し、本剤の使用実態に関する情報(患者背景、有効性・安全性(他剤併用時の有効性・安全性を含む)及び薬物相互作用のデータ等)を収集して定期的に報告するとともに、調査の結果を再審査申請時に提出すること。
22. 包装
30錠[瓶、バラ、乾燥剤入り]
28錠[7錠(PTP)×4、乾燥剤入り]
**,*23. 主要文献
- Zash R et al. N Engl J Med 2018; 379(10): 979-81.
- Bonaboud S, et al. Antimicrob Agents Chemother 2011; 55(3): 1315-7.
- BIC-in vitro transporters inhibition: AD-141-2285(承認年月日: 2019年3月26日, CTD 2.6.4.7)
- BIC-in vitro metabolism by CYP: AD-141-2290(承認年月日: 2019年3月26日, CTD 2.6.4.7)
- BIC-in vitro metabolism by UGT: AD-141-2291(承認年月日: 2019年3月26日, CTD 2.6.4.7)
- FTC-Mass balance study: FTC-106(承認年月日: 2019年3月26日, CTD 2.7.2.3, 2.6.4.5, 2.7.6.1)
- In vitro DDI study with transporters: AD-120-2018(承認年月日: 2019年3月26日, CTD 2.6.4.7, 2.6.5.11)
- Japanese ethnic sensitivity study: GS-US-380-1991(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.6.1)
- BIC-Population PK analysis(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.3)
- FTC-PK analysis(承認年月日: 2019年3月26日, CTD 2.7.2.3)
- TAF-Population PK analysis(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.3)
- Food effect: GS-US-141-1233(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.6.1)
- BIC-In vitro protein binding: AD-141-2287(承認年月日: 2019年3月26日, CTD 2.6.4.4, 2.7.2.3)
- BIC-Blood to plasma ratio: AD-141-2312(承認年月日: 2019年3月26日, CTD 2.6.4.4, 2.7.2.3)
- FTC-In vitro protein binding: TBZZ/93/0025(承認年月日: 2019年3月26日, CTD 2.6.4.4, 2.6.5.6, 2.7.2.3)
- TAF-Examination of pharmacokinetics: GS-US-120-0108/0114(承認年月日: 2019年3月26日, CTD 2.6.4.4, 2.7.2.3)
- In vitro human plasma protein binding: P0504-00039.1(承認年月日: 2019年3月26日, CTD 2.6.4.4, 2.6.5.6)
- Birkus G et al. Antimicrob Agents Chemother 2007;51(2):543-50.
- Birkus G et al. Mol Pharmaco2008; 74(1):92-100.
- Eisenberg EJ et al. Nucleosides Nucleotides Nucleic Acids 2001;20(4-7):1091-8.
- BIC-Mass balance study: GS-US-141-1481(承認年月日: 2019年3月26日, CTD 2.7.2.2, 2.7.2.3)
- TAF-Mass Balance study: GS-US-120-0109(承認年月日: 2019年3月26日, CTD 2.7.2.3)
- BIC-Hepatic impairment: GS-US-141-1478(承認年月日: 2019年3月26日, CTD 2.7.2.3)
- TAF-Hepatic impairment: GS-US-120-0114(承認年月日: 2019年3月26日, CTD 2.6.4.4, 2.7.2.3)
- TAF-Hepatic impairment: GS-US-320-1615(承認年月日: 2019年3月26日, CTD 2.7.2.3)
- BIC-Renal impairment: GS-US-141-1479(承認年月日: 2019年3月26日, CTD 2.7.2.2, 2.7.2.3)
- FTC-Renal impairment: FTC-107(承認年月日: 2019年3月26日, CTD 2.7.2.3)
- TAF-Renal impairment: GS-US-120-0108(承認年月日: 2019年3月26日, CTD 2.6.4.4, 2.7.2.3)
- TAF-Population PK in patients with dialysis and ESRD: QP 2015-1004 TAF ESRD(承認年月日: 2016年12月19日, Vemlidy CTD 2.7.2.3)
- FTC-In vitro Interaction study: AD-236-2010(承認年月日: 2019年3月26日, CTD 2.6.4.7, 2.6.5.11)
- BIC DDI study: GS-US-141-1485(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.2.3)
- BIC/F/TAF-DDI study: GS-US-380-1761(承認年月日: 2019年3月26日 CTD 2.7.2.1, 2.7.2.2, 2.7.2.3)
- BIC/F/TAF-DDI study: GS-US-380-1999(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.2.3)
- BIC/F/TAF-DDI study: GS-US-380-3909(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.2.3)
- DESCOVY-DDI study: GS-US-311-1387(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.2.3)
- BIC/F/TAF-DDI study: GS-US-380-3908(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.2.3)
- BIC-Drug interaction potential: GS-US-380-4270(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.2.3)
- DDI study with: GS-US-311-1790(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.2.3)
- GENVOYA-DDI study: GS-US-292-1316(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.2, 2.7.2.3)
- Phase 3 study: GS-US-380-1489(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.3, 2.7.2.4, 2.7.3.2, 2.7.3.3, 2.7.4.2, 2.7.6.1)
- Phase 3 study: GS-US-380-1490(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.3, 2.7.2.4, 2.7.3.2, 2.7.3.3, 2.7.4.2, 2.7.6.1)
- Phase 3 study: GS-US-380-1844(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.3, 2.7.2.4, 2.7.3.2, 2.7.3.3, 2.7.4.2, 2.7.6.1)
- Phase 3 study: GS-US-380-1878(承認年月日: 2019年3月26日, CTD 2.7.2.1, 2.7.2.3, 2.7.2.4, 2.7.3.2, 2.7.3.3, 2.7.4.2, 2.7.6.1)
- QT/QTc interval study: GS-US-141-1480(承認年月日: 2019年3月26日, CTD 2.7.2.2, 2.7.2.3, 2.7.6.1)
- QT/QTc interval study: GS-US-120-0107(承認年月日: 2019年3月26日, CTD, 2.7.2.1, 2.7.2.3, 2.7.6.1)
- BIC-In vitro antiviral activity: PC-141-2032(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.2.3, 2.6.6.3, 2.6.7.7)
- BIC-In vitro antiviral activity: PC-141-2034(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.2.3, 2.6.6.6, 2.6.7.11, 2.6.7.13)
- BIC-In vitro antiviral activity: PC-141-2036(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.2.3, 2.6.6.6, 2.6.7.11, 2.6.7.13)
- BIC-In vitro antiviral activity: PC-141-2043(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.2.3)
- Paff MT, et al. Antimicrob Agents Chemother 1994; 38(6): 1230-8.
- George RP, et al. Drugs Future 1995; 20(8): 761-5.
- Feng JY et al. FASEB J. 1999;13(12):1511-17
- Robbins BL et al. Pharmacotherapy 2003;23(6):695-701.
- Anti-virus activity against human and animal virus: PC-120-2003(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.4.7, 2.6.5.11)
- Robbins BL et al. Antimicrob. Agents Chemother. 1998:42(3):612-17
- Cihlar T et al. Antivir. Chem. Chemother. 1997:8(3):187-95
- BIC-In vitro antiviral activity: PC-141-2057(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.2.3)
- BIC-In vitro antiviral activity: PC-141-2035(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.2.3)
- Schinazi RF, et al. Antimicrob Agents Chemother 1992;36(11):2423-31.
- Painter G, et al. Anti-HIV, Anti-Hepatitis B Virus. Drugs of the Future 1995;20(8):761-5.
- Jeong LS, et al. J Med Chem 1993;36(2):181-95.
- TAF-In vitro antiviral activity: PC-120-2003/2004/2007/2017(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.4.7, 2.6.5.11)
- BIC- In vitro antiviral activity: PC-141-2052(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.3.1)
- BIC- In vitro antiviral activity: PC-141-2056(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.6.3.1)
- Ntemgwa M, et al. Antimicrob Agents Chemother 2009;53(2):708-15.
- Kagan RM, et al. Antiviral Res 2007;75(3):210-8.
- BIC-In vitro antiviral activity: PC-141-2055(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.7.2.2)
- BIC-In vitro antiviral activity: PC-141-2040(承認年月日: 2019年3月26日, CTD 2.6.2.2, 2.7.2.2)
- Schinazi RF, et al. Antimicrob Agents Chemother 1993;37(4):875-81.
- TAF-In vitro antiviral activity: PC-120-2011(承認年月日: 2019年3月26日, CTD 2.6.4.4, 2.6.5.5, 2.6.6.7, 2.6.7.16)
- Margot NA, et al. Antimicrob Agents Chemother 2006;50(12):4087-95
- TAF-In vitro antiviral activity: PC-120-2015(承認年月日: 2019年3月26日, CTD 2.6.2.2)
- E/C/F/TAF, BIC/F/TAF ESRD: GS-US-292-1825
- BIKTARVY 米国添付文書
- Eron J et.al. Lancet HIV 2019; 6: e15-24
- Phase 3 study: GS-US-380-4030
24. 文献請求先及び問い合わせ先
ギリアド・サイエンシズ株式会社
メディカルサポートセンター
〒100-6616 東京都千代田区丸の内一丁目9番2号
グラントウキョウサウスタワー
フリーダイアル 0120-506-295
FAX 03-5958-2959
受付時間:9:00~17:30(土・日・祝日及び会社休日を除く)
26. 製造販売業者等
26.1 製造販売元
ギリアド・サイエンシズ株式会社
〒100-6616 東京都千代田区丸の内一丁目9番2号
グラントウキョウサウスタワー