レイアタッツカプセル150mg/200mgの添付文書
- 商品名:
- レイアタッツカプセル150mg/200mg
- 一般名:
- アタザナビル
- 略称 :
- ATV

添付文書の読み方
ここで提供している添付文書情報は、2021年12月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
HIVプロテアーゼ阻害剤
アタザナビル硫酸塩カプセル
- **2021年5月改訂(第2版)
*2021年1月改訂 - 劇薬,処方箋医薬品注)
注)注意-医師等の処方箋により使用すること
| 日本標準商品分類番号 | 87625 |
|---|
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 2年 |
| 150mg | 200mg | |
|---|---|---|
| 承認番号 | 21500AMY00158000 | 21500AMY00159000 |
| 販売開始 | 2004年1月 | 2004年1月 |
2. 禁忌(次の患者には投与しないこと)
2.1
本剤の成分に対し過敏症の既往歴のある患者
2.2
重度の肝障害のある患者[9.3.1参照]
*2.3
次の薬剤を投与中の患者:リファンピシン,イリノテカン塩酸塩水和物,ミダゾラム,トリアゾラム,ベプリジル塩酸塩水和物,エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン,ジヒドロエルゴタミンメシル酸塩,エルゴメトリンマレイン酸塩,メチルエルゴメトリンマレイン酸塩,ピモジド,シンバスタチン,ロバスタチン(国内未発売),ロミタピドメシル酸塩,バルデナフィル塩酸塩水和物,ブロナンセリン,アスナプレビル,アゼルニジピン,オルメサルタン メドキソミル・アゼルニジピン,ルラシドン塩酸塩,リバーロキサバン,リオシグアト,グラゾプレビル水和物,グレカプレビル水和物・ピブレンタスビル,プロトンポンプ阻害剤(オメプラゾール,ランソプラゾール,ラベプラゾール,エソメプラゾール,ボノプラザンフマル酸塩),アスピリン・ランソプラゾール,アスピリン・ボノプラザンフマル酸塩,セイヨウオトギリソウ(St. John’s Wort, セント・ジョーンズ・ワート)[10.1参照]
3. 組成・性状
3.1 組成
| 販売名 | レイアタッツカプセル150mg | レイアタッツカプセル200mg |
|---|---|---|
| 有効成分 | 1カプセル中 アタザナビル硫酸塩 170.84mg (アタザナビルとして150mgに相当) |
1カプセル中 アタザナビル硫酸塩 227.79mg (アタザナビルとして200mgに相当) |
| 添加剤 | クロスポビドン,乳糖水和物及びステアリン酸マグネシウム また,カプセル本体にゼラチン及び青色二号 |
クロスポビドン,乳糖水和物及びステアリン酸マグネシウム また,カプセル本体にゼラチン及び青色二号 |
3.2 製剤の性状
| 販売名 | 色 | 内容物 | 形状 | サイズ | 識別コード (印字色) |
|---|---|---|---|---|---|
| レイアタッツカプセル150mg | キャップ:青色 ボディ:淡青色 |
白色~淡黄色の顆粒 |  |
1号 カプセル |
BMS 150mg(白色) 3624(青色) |
| レイアタッツカプセル200mg | キャップ:青色 ボディ:青色 |
白色~淡黄色の顆粒 |  |
0号 カプセル |
BMS 200mg(白色) 3631(白色) |
4. 効能又は効果
HIV-1感染症
6. 用法及び用量
通常,成人には以下の用法・用量に従い食事中又は食直後に経口投与する。
投与に際しては必ず他の抗HIV薬と併用すること。
〈抗HIV薬による治療経験のない患者〉
- アタザナビルとして300mgとリトナビルとして100mgをそれぞれ1日1回併用投与
- アタザナビルとして400mgを1日1回投与
〈抗HIV薬による治療経験のある患者〉
- アタザナビルとして300mgとリトナビルとして100mgをそれぞれ1日1回併用投与
7. 用法・用量に関連する注意
7.1
リトナビル100mgを超えて併用投与した際の有効性と安全性は確立していない。リトナビルを高用量で併用投与した場合には本剤の安全性プロファイル(心伝導障害,高ビリルビン血症)に影響をあたえる可能性がある。
7.2
ウイルス学的治療失敗を伴う抗HIV薬による治療経験のある患者に,本剤をリトナビルと併用せずに投与することは推奨されない。[17.1.3,17.1.4参照]
7.3
抗HIV薬による治療経験のない患者で,リトナビルの投与が適用できない患者に対しては,リトナビルと併用しない用法・用量(アタザナビルとして400mgを1日1回投与)を考慮すること。[17.1.1,17.1.2参照]
7.4
中等度の肝障害患者(Child-Pugh分類B)には,リトナビルを併用せずに,本剤の投与量を300mg,1日1回に減量して投与することを考慮する。中等度の肝障害のある患者には,本剤とリトナビルの併用は推奨されない。[9.3.3,16.6.2参照]
7.5
透析を施行している腎障害患者の場合,抗HIV薬による治療経験のない患者には,本剤をリトナビルと併用して投与すること。なお,抗HIV薬による治療経験のある患者には,本剤を投与しないこと。[9.2.1,16.6.1参照]
7.6
本剤と他の抗HIV薬との併用療法において,因果関係が特定できない重篤な副作用が発現し,治療の継続が困難であると判断された場合には,原則として本剤及び併用している他の抗HIV薬の投与をすべて一旦中止すること。
7.7
ヒト免疫不全ウイルス(HIV)は感染初期から多種多様な変異株を生じ,薬剤耐性を発現しやすいことが知られているので,本剤は他の抗HIV薬と併用すること。
7.8
本剤の減量投与に対する長期的な有効性は確立されていないので,本剤を減量して投与することは推奨されない。
7.9
本剤とテノホビルを併用する場合,本剤300mg,リトナビル100mg,テノホビル300mgをそれぞれ1日1回食事中又は食直後に投与することが推奨される。[10.2参照]
8. 重要な基本的注意
**8.1
本剤の使用に際しては,国内外のガイドライン等の最新の情報を参考に,患者又はそれに代わる適切な者に,次の事項についてよく説明し同意を得た後,使用すること。
8.1.1
本剤はHIV-1感染症の根治療法薬ではないことから,日和見感染を含むHIV-1感染症の進展に伴う疾病を発症し続ける可能性があるので,本剤投与開始後の身体的状況の変化については,すべて担当医に報告すること。
8.1.2
本剤を空腹時に服用すると血中濃度が低くなり抗ウイルス作用を発揮できないことがあるため,本剤を食事中又は食直後に服用すること。
8.1.3
本剤投与開始後,担当医の指示なしに用量を変更したり,服用を中止したりせず,処方された用量を守ること。
8.1.4
本剤は一部の薬剤と相互作用を起こすことがあるため,処方箋の有無にかかわらず服用している薬剤をすべて担当医及び薬剤師に報告すること。
**8.1.5
抗HIV療法による効果的なウイルス抑制は,性的接触による他者へのHIV感染の危険性を低下させることが示されているが,その危険性を完全に排除することはできないこと。
**8.1.6
抗HIV療法が,血液等による他者へのHIV感染の危険性を低下させるかどうかは証明されていないこと。
8.1.7
無症候性の高ビリルビン血症があらわれることがあるので,本剤服用中に眼球・皮膚の黄染がみられた場合には担当医に報告すること。
8.1.8
本剤の長期投与による影響については,現在のところ不明であること。
8.2
本剤にて治療中,UDP-グルクロニルトランスフェラーゼ(UGT)阻害により無症候性の非抱合型ビリルビン上昇が高頻度にあらわれる。この高ビリルビン血症は本剤投与中止により回復する。高ビリルビン血症とともに肝トランスアミナーゼの上昇を認める場合には,他の原因を疑うこと。総ビリルビンの正常範囲の上限より5倍を超える上昇が認められた患者での長期的な安全性データは得られていない。ビリルビン上昇による黄疸・黄疸眼があらわれ,患者の美容上の観点より,本剤から他の抗HIV療法への切り換えを考慮することがある。
8.3
本剤の投与による軽・中等度の発疹が報告されている。一般に投与開始3週間以内に斑状又は丘疹状の発疹が生じ,通常は投与継続中に2週間以内で消失する。重度の発疹が発現したり,持続する場合には本剤の投与を中止すること。[11.1.5参照]
8.4
HIVプロテアーゼ阻害薬にて治療中の患者において糖尿病の発症や悪化及び高血糖が発現し,その中には糖尿病性ケトアシドーシスを伴っていた症例が市販後調査で報告されている。定期的に検査を行うなど観察を十分に行うこと。[11.1.2参照]
8.5
本剤と乳酸アシドーシスの危険性を増大させることが知られているヌクレオシドアナログを併用投与した患者(妊婦を含む)に,致死性の乳酸アシドーシス及び高乳酸血症が報告されている。
8.6
本剤を含む抗HIV薬の多剤併用療法を行った患者で,免疫再構築症候群が報告されている。投与開始後,免疫機能が回復し,症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス,サイトメガロウイルス,ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また,免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症,多発性筋炎,ギラン・バレー症候群,ブドウ膜炎等)が発現するとの報告があるので,これらの症状を評価し,必要時には適切な治療を考慮すること。
8.7
重度の肝機能障害,肝炎等があらわれることがあるので,定期的に肝機能検査を行うなど観察を十分に行うこと。[9.1.4,9.3.2,9.3.3,11.1.1参照]
8.8
本剤による治療は,抗HIV療法に十分な経験を持つ医師のもとで開始すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 心伝導障害(房室ブロック)のある患者
本剤の投与により,心電図検査でPR間隔の延長を示すことがある。心伝導障害は第一〜三度AVブロックの報告がある。臨床試験データが十分でない。[10.2,11.1.4,17.3.1参照]
9.1.2 血友病及び著しい出血傾向を有する患者
HIVプロテアーゼ阻害薬にて治療中の血友病患者において突発性の出血性関節症をはじめとする出血事象の増加が報告されている。[11.1.3参照]
9.1.3 無酸症等著しい低胃酸状態が持続する状態の患者
無酸症等著しい低胃酸状態が持続する状態では,本剤の血中濃度が低下し作用が減弱するおそれがある。
9.1.4 B型・C型肝炎の患者
定期的に肝機能検査を行うなど患者の状態をモニタリングすること。トランスアミナーゼがさらに上昇する又は肝機能が悪化するおそれがある。[8.7参照]
9.2 腎機能障害患者
9.2.1 透析を施行している腎障害患者
[7.5,16.6.1参照]
9.3 肝機能障害患者
9.3.1 重度の肝障害のある患者
投与しないこと。血中濃度が上昇すると予想される。[2.2,16.6.2参照]
9.3.2 投与前に著しいトランスアミナーゼの上昇が認められた患者
定期的に肝機能検査を行うなど患者の状態をモニタリングすること。トランスアミナーゼがさらに上昇する又は肝機能が悪化するおそれがある。[8.7参照]
9.3.3 軽度~中等度の肝障害のある患者
本剤は主に肝臓で代謝されるため,肝障害のある患者では高い血中濃度が持続するおそれがある。[7.4,8.7,16.6.2参照]
9.5 妊婦
9.5.1
妊婦又は妊娠している可能性のある女性には,治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験(ラット,ウサギ)では,母動物の曝露量が臨床用量(400mg/日)と同程度(ウサギ)又は2倍(ラット)で催奇形性は認められなかった。ラットの周産期及び授乳期に投与すると,母動物に毒性が発現する用量(曝露量で臨床用量の2倍に相当)で,産児に体重減少又は体重増加抑制が認められた。母動物の曝露量がヒトに400mg/日投与した場合の曝露量と同程度の用量では,産児に対する影響は認められなかった。
9.5.2
分娩前に追加検査及び代替治療の実施を考慮すること。
本剤投与中に高ビリルビン血症が高頻度に発現する。本剤を妊婦に投与した場合,新生児や乳幼児に生理的高ビリルビン血症の悪化及び核黄疸の発現がみられるか否かは不明である。
9.6 授乳婦
授乳を避けさせること。乳汁を介してHIV母児感染の可能性があること及び本剤の乳汁中への移行により乳児に重篤な有害事象が発現する可能性がある。動物実験(ラット)で,乳汁中に移行することが報告されている。また,本剤がヒトの乳汁中に移行するとの報告がある。
9.7 小児等
9.7.1
新生児,月齢3ヵ月未満の乳児には,核黄疸の発現の危険性があるので本剤を投与しないこと。[17.1.5参照]
9.7.2
小児等に対する国内臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能(肝機能,腎機能,心機能等)が低下しており,また,合併症を有し,若しくは他の薬剤を併用している場合が多い。[16.6.3参照]
10. 相互作用
本剤はチトクロームP450(CYP3A4)及びUDP-グルクロニルトランスフェラーゼ(UGT)の阻害作用を有する。[16.7参照]
10.1 併用禁忌(併用しないこと)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| リファンピシン(リファジン) [16.7参照] |
本剤の血中濃度が低下し,本剤の効果が減弱するおそれがある。 | リファンピシンがCYP3A4を誘導することによる。 |
| イリノテカン塩酸塩水和物 (カンプト,トポテシン) |
イリノテカンの副作用を増強することがある。 | 本剤のUGT阻害によりイリノテカンの代謝が抑制されるおそれがある。 |
| ミダゾラム(ドルミカム) トリアゾラム(ハルシオン) |
これらの薬剤の代謝が抑制され,重篤な又は生命に危険を及ぼすような事象(持続的又は過度の鎮静,呼吸抑制等)が起こる可能性がある。 | CYP3A4に対する競合による。 |
| ベプリジル塩酸塩水和物(ベプリコール) | 重篤な又は生命に危険を及ぼすような事象が起こる可能性がある。 | |
| エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン(クリアミン配合錠) ジヒドロエルゴタミンメシル酸塩 エルゴメトリンマレイン酸塩(エルゴメトリンF) メチルエルゴメトリンマレイン酸塩(パルタン) |
これらの薬剤の代謝が抑制され,重篤な又は生命に危険を及ぼすような事象(末梢血管収縮,四肢の虚血等を特徴とする急性の毒性作用)が起こる可能性がある。 | |
| ピモジド(オーラップ) | ピモジドの代謝が抑制され,重篤な又は生命に危険を及ぼすような事象(不整脈等)が起こる可能性がある。 | |
| シンバスタチン(リポバス) ロバスタチン(国内未発売) |
これらの薬剤の代謝が抑制され,重篤な又は生命に危険を及ぼすような事象(横紋筋融解症を含むミオパチー等)が起こる可能性がある。 | |
| ロミタピドメシル酸塩(ジャクスタピッド) | 本剤によりロミタピドの血中濃度が著しく上昇するおそれがある。 | |
| バルデナフィル塩酸塩水和物(レビトラ) | 本剤との併用に関する試験は行われていないが,バルデナフィルの血中濃度が上昇し,有害事象(低血圧,視覚障害,持続勃起症,失神等)の発現が増加するおそれがある。 | |
| ブロナンセリン(ロナセン) | 本剤によりブロナンセリンの血中濃度が上昇し,作用が増強するおそれがある。 | |
| *アスナプレビル(スンベプラ) | アスナプレビルの血中濃度が上昇する。肝臓に関連した有害事象が発現し,また重症化するおそれがある。 | |
| *アゼルニジピン(カルブロック) オルメサルタンメドキソミル・アゼルニジピン(レザルタス) |
本剤によりアゼルニジピンの血中濃度が上昇し,作用が増強するおそれがある。 | |
| *ルラシドン塩酸塩(ラツーダ) | 本剤によりルラシドンの血中濃度が上昇し,作用が増強するおそれがある。 | |
| リバーロキサバン(イグザレルト) | 本剤によりリバーロキサバンの血中濃度が上昇し,作用が増強するおそれがある。 | CYP3A4及びP糖蛋白(P-gp)の強力な阻害作用によりリバーロキサバンのクリアランスが減少する。 |
| リオシグアト(アデムパス) | ケトコナゾールとの併用によりリオシグアトの血中濃度が上昇し,クリアランスが低下したとの報告がある。 | 複数のCYP分子種(CYP1A1, CYP3A等)及びP-gp/乳癌耐性蛋白(BCRP)阻害によりリオシグアトのクリアランスが低下する。 |
| グラゾプレビル水和物(グラジナ) | グラゾプレビルの血中濃度が上昇するおそれがある。 | 本剤のOATP1Bに対する阻害作用によるものと考えられている。 |
| グレカプレビル水和物・ピブレンタスビル(マヴィレット配合錠) | グレカプレビルの血中濃度が上昇するおそれがある。 ALT上昇のリスクが増加するおそれがある。 |
本剤のOATP1Bに対する阻害作用によるものと考えられている。 ALT上昇の機序は不明。 |
| *プロトンポンプ阻害剤 オメプラゾール(オメプラール,オメプラゾン) ランソプラゾール(タケプロン) ラベプラゾール(パリエット) エソメプラゾール(ネキシウム) ボノプラザンフマル酸塩(タケキャブ) アスピリン・ランソプラゾール(タケルダ) アスピリン・ボノプラザンフマル酸塩(キャブピリン) [16.7参照] |
本剤とこれら薬剤の併用により,血中濃度が低下し,本剤の効果が減弱するおそれがある。 | 本剤の溶解性がpHに依存することから,胃酸分泌抑制により本剤の吸収が抑制されるおそれがある。 |
| セイヨウオトギリソウ(St. John’s Wort,セント・ジョーンズ・ワート)含有食品 | 本剤の代謝が促進され血中濃度が低下するおそれがあるので,本剤投与時はセイヨウオトギリソウ含有食品を摂取しないよう注意すること。 | セイヨウオトギリソウにより誘導された肝薬物代謝酵素(チトクロームP450)が本剤の代謝を促進し,クリアランスを上昇させるためと考えられている。 |
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| テノホビル ジソプロキシルフマル酸塩[7.9,16.7参照] | 本剤のAUC,Cminが低下し,テノホビルの血中濃度が上昇するおそれがある。テノホビルに関連した有害事象(腎障害等)を増強するおそれがあるので,併用する場合にはテノホビルに関連した有害事象のモニタリングを行うこと。 リトナビルを併用しない場合には,本剤とテノホビルの併用は推奨されない。 |
機序不明 |
| エファビレンツ [16.7参照] |
本剤とエファビレンツの併用は推奨されない。 | 本剤の血中濃度が低下するおそれがある。 |
| ネビラピン[16.7参照] | 本剤の血中濃度が低下し,ネビラピンの血中濃度が上昇するおそれがある。本剤とネビラピンの併用は推奨されない。 | ネビラピンがCYP3A4を誘導し,また代謝が阻害されることによる。 |
| ホスアンプレナビルカルシウム水和物 | ホスアンプレナビル700mg/リトナビル100mg1日2回と本剤300mg1日1回を併用した場合,本剤のCmax,AUCはそれぞれ24%,22%減少した。 | CYP3A4に対する競合による。 |
| 制酸剤,緩衝作用を有する薬剤 乾燥水酸化アルミニウムゲル,沈降炭酸カルシウム等 |
本剤はこれらの薬剤投与の2時間前又は1時間後に投与すること。 | 本剤の吸収が抑制されるおそれがある。 |
| H2受容体拮抗剤 ファモチジン等[16.7参照] |
本剤とこれら薬剤の併用により,本剤の血中濃度が著しく低下し,効果が減弱するおそれがある。H2受容体拮抗剤の影響を減少させるために,必ず本剤とリトナビルを併用して投与し,本剤とH2受容体拮抗剤は可能な限り間隔をあけて投与することが推奨される。 また,抗HIV薬による治療経験のある患者に,本剤/リトナビルとテノホビルを併用する場合は,H2受容体拮抗剤の併用は推奨されない。 |
胃内pHの上昇により,本剤の吸収が抑制されるおそれがある。 |
| アミオダロン キニジン リドカイン 三環系抗うつ薬 |
本剤とこれら薬剤の併用により重篤な又は生命に危険を及ぼすような有害事象があらわれるおそれがあり,この併用に関する試験は行われていない。併用する場合には,これらの薬剤の血中濃度のモニタリングを行うことが望ましい。 | これらの薬剤の血中濃度が上昇するおそれがある。 |
| トラゾドン | トラゾドンの血中濃度が上昇するおそれがある。併用する場合には,患者の状態に注意し,必要に応じてトラゾドンの減量を考慮すること。 | 本剤がCYP3A4を阻害する。 |
| リファブチン [16.7参照] |
リファブチンの作用が増強するおそれがあるので,リファブチンの用法・用量を150mg隔日投与又は1週間に3回投与とすることが推奨される。併用する場合には,副作用のモニタリングを十分に行うこと。 | 本剤がCYP3A4を阻害することにより,リファブチンの血中濃度を上昇させる。 |
| ワルファリン | 本剤との併用により重篤な又は生命に危険を及ぼすような出血があらわれるおそれがあり,この併用に関する試験は行われていない。併用する場合には,INRのモニタリングを行うことが望ましい。 | ワルファリンの血中濃度が上昇するおそれがある。 |
| ジルチアゼム [9.1.1,16.7,17.3.1参照] |
本剤(400mg1日1回)とジルチアゼム(180mg1日1回)を併用した場合にジルチアゼム及びデスアセチル-ジルチアゼムのCmax,AUCが約2〜3倍に増加するとの報告がある。ジルチアゼムを半量に減量して投与することを考慮すること。本剤の投与により,心電図検査でPR間隔の延長を示すことがある。併用する場合には心電図のモニタリングを行うことが望ましい。 | ジルチアゼム及びデスアセチル-ジルチアゼムの血中濃度が上昇するおそれがある。 |
| フェロジピン ニフェジピン ニカルジピン ベラパミル |
フェロジピン,ニフェジピン,ニカルジピンあるいはベラパミルと本剤を併用する場合にはこれらの薬剤を減量するなど用量に注意すること。併用する場合には心電図のモニタリングを行うことが望ましい。 | これらの薬剤の血中濃度が上昇するおそれがある。 |
| シルデナフィルクエン酸塩 タダラフィル |
これらの薬剤の血中濃度が上昇し,有害事象(低血圧,視覚障害,持続勃起症,失神等)を起こすおそれがある。併用する場合には,有害事象のモニタリングを行うなど注意すること。 | CYP3A4に対する競合による。 |
| アトルバスタチン ロスバスタチン |
これらの薬剤の血中濃度が上昇するおそれがある。 本剤を含むHIVプロテアーゼ阻害薬とこれらの薬剤を併用した場合,横紋筋融解症を含むミオパチー等の事象発現の危険性が高くなる可能性があるので,注意すること。 |
|
| シクロスポリン タクロリムス |
併用する場合には,治療域のモニタリングを行うことが望ましい。 | これらの薬剤の血中濃度が上昇するおそれがある。 |
| テムシロリムス | テムシロリムス及びその活性代謝物であるシロリムスの血中濃度が上昇するおそれがある。 | CYP3A4に対する阻害による。 |
| クラリスロマイシン [16.7参照] |
本剤(400mg1日1回)とクラリスロマイシン(500mg1日1回)を併用した場合にクラリスロマイシンのCmaxが約1.5倍,AUCが約2倍に増加するとの報告がある。クラリスロマイシンに関連する有害事象(QTc延長等)を起こすおそれがあるので,クラリスロマイシンを半量に減量して投与することを考慮すること。また,活性代謝物である14位水酸化体の濃度が顕著に低下するとの報告があり,Mycobacteriumavium complexによる感染症以外の症状に対しては代替の治療法を考慮すること。 | 本剤及びクラリスロマイシンの血中濃度が上昇するおそれがある。 |
| ブプレノルフィン塩酸塩 | ブプレノルフィンの血中濃度が上昇するおそれがある。本剤/リトナビルと併用する場合は,鎮静状態及び認知機能のモニタリングを行い,ブプレノルフィンの減量を考慮すること。また,リトナビルを併用しない場合には,本剤の血中濃度が減少するおそれがあるので,本剤とブプレノルフィンの併用は推奨されない。 | 本剤がCYP3A4及びUGT1A1を阻害する。 |
| エチニルエストラジオール及びノルエチステロン又はノルゲスチメートを含む経口避妊薬 [16.7参照] |
本剤/リトナビルと併用する場合は,エチニルエストラジオールとして0.035mg以上の経口避妊薬を投与することが望ましい。 また,リトナビルを併用せずに本剤と併用する場合は,エチニルエストラジオールとして0.030mg以下の経口避妊薬を投与することが望ましい。 黄体ホルモン薬の血中濃度上昇による長期的な影響は不明であるが,インスリン抵抗性,脂質異常症,ざ瘡のリスクを上昇させるおそれがあるので,注意すること。本剤投与時は他の避妊法を行うことが望ましい。 |
本剤/リトナビルとエチニルエストラジオール及びノルゲスチメートを含む経口避妊薬の併用により,エチニルエストラジオールの平均血中濃度が低下し,17-デアセチルノルゲスチメートの平均血中濃度が上昇するおそれがある。 本剤(リトナビルの併用なし)とエチニルエストラジオール及びノルエチステロンを含む経口避妊薬の併用により,エチニルエストラジオール及びノルエチステロンの平均血中濃度が上昇するおそれがある。 |
| エトラビリン | 本剤の血中濃度が減少し,エトラビリンの血中濃度が上昇するおそれがある。 | CYP3A4誘導作用により,本剤の代謝が促進される。また,本剤のCYP3A4阻害作用により,エトラビリンの代謝が阻害される。 |
| マラビロク,ダサチニブ水和物 | これらの薬剤の血中濃度が上昇するおそれがある。 | 本剤がCYP3A4の活性を阻害する。 |
| ケトコナゾール(国内未発売) イトラコナゾール [16.7参照] |
本剤/リトナビルとケトコナゾール又はイトラコナゾールを併用する場合は,注意すること。 | これらの薬剤はCYP3A4を阻害し,またCYP3A4により代謝される。 |
| ボリコナゾール [16.7参照] |
CYP2C19の活性型遺伝子を1つ以上有する患者(Extensive Metabolizer:EM)注)に本剤/リトナビル(300mg/100mg1日1回)とボリコナゾール(200mg1日2回)を併用した場合,ボリコナゾール及び本剤の血漿中濃度が低下するおそれがある。一方,CYP2C19の活性型遺伝子を有さない患者(Poor Metabolizer:PM)注)に本剤/リトナビル(300mg/100mg1日1回)とボリコナゾール(50mg1日2回)を併用した場合,ボリコナゾールの血漿中濃度が上昇し,本剤の血漿中濃度が低下するおそれがある。 併用する場合には,ボリコナゾールに関連した有害事象,及びボリコナゾールあるいは本剤の有効性の減弱について注意深く観察すること。 |
CYP2C19のEMでは,リトナビルが,ボリコナゾールの主な肝薬物代謝酵素であるCYP2C19を誘導することにより,ボリコナゾールの血漿中濃度が低下する。 CYP2C19のPMでは,リトナビル及び本剤が,CYP3A4による代謝を阻害することにより,ボリコナゾールの血漿中濃度が上昇する。 本剤の血漿中濃度が低下する機序は不明である。 |
| CYP3A4の基質となる薬剤 ボセンタン水和物等 |
これらの薬剤の血中濃度が上昇するおそれがある。 | 本剤がCYP3A4を阻害する。 |
| エルバスビル | エルバスビルの血中濃度が上昇するおそれがある。 | CYP3A4に対する競合による。 |
注):CYP2C19遺伝子型
EM:CYP2C19 *1/*1,CYP2C19 *1/*2,CYP2C19 *1/*3,CYP2C19 *1/*17,CYP2C19 *2/*17,CYP2C19 *3/*17,CYP2C19 *17/*17
PM:CYP2C19 *2/*2,CYP2C19 *2/*3,CYP2C19 *3/*3
11. 副作用
次の副作用があらわれることがあるので,観察を十分に行い,異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1 重大な副作用
11.1.1 重度の肝機能障害,肝炎
重度の肝機能障害,肝炎(1%未満)等があらわれることがある。[8.7参照]
11.1.2 糖尿病,糖尿病の悪化及び高血糖
[8.4参照]
11.1.3 出血傾向
出血事象があらわれた場合には血液凝固因子を投与するなど適切な処置を行うこと。[9.1.2参照]
11.1.4 QT延長,心室頻拍(torsades de pointesを含む),房室ブロック(第一度〜第三度AVブロック)
[9.1.1参照]
11.1.5 皮膚粘膜眼症候群(Stevens-Johnson症候群),多形紅斑,中毒性皮疹
[8.3参照]
11.1.6 尿細管間質性腎炎
腎間質に結晶の沈着が認められた症例が報告されている。
11.2 その他の副作用
| 1%以上 | 1%未満 | 頻度不明 | |
|---|---|---|---|
| 心臓障害 | 失神,浮腫,動悸,心停止,第一度AVブロック,心筋炎 | QT延長,torsades de pointes | |
| 神経系障害 | 頭痛(2.7%) | 末梢神経障害,健忘,傾眠,浮動性めまい,味覚異常,灼熱感,痙攣,運動過多,感覚鈍麻,反射亢進 | |
| 眼障害 | 黄疸眼(1.1%) | ||
| 耳及び迷路障害 | 耳鳴,耳炎 | ||
| 呼吸器,胸郭及び縦隔障害 | 呼吸困難,咳嗽,しゃっくり,低酸素症 | ||
| 胃腸障害 | 悪心(5.7%),嘔吐(2.2%),下痢(2.2 %),消化不良(1.2%),腹痛(1.1%) | 口渇,鼓腸,胃炎,膵炎,アフタ性口内炎,腹部膨満,大腸炎,便秘,歯痛,食道潰瘍,食道炎,胃腸炎,胃腸障害,口腔内潰瘍形成,消化性潰瘍 | |
| 腎及び尿路障害 | 血尿,頻尿,蛋白尿,腎結石,腎臓痛,尿異常,結晶尿,腎不全,乏尿,多尿,尿路感染 | ||
| 皮膚及び皮下組織障害 | 発疹(1.9%) | 脱毛症,そう痒症,蕁麻疹,血管拡張,水疱性皮膚炎,湿疹,血管浮腫,脂肪萎縮(顔面),光線過敏,多汗,斑状出血,紫斑,蜂巣炎,皮膚糸状菌症,皮膚乾燥,爪の障害,脂漏 | |
| 筋骨格系及び結合組織障害 | 関節痛,筋萎縮,筋肉痛,ミオパチー,背部痛,骨痛,四肢痛,筋無力症,ピクピクした動き | ||
| 代謝及び栄養障害 | 食欲不振,食欲亢進,体重減少,体重増加,脱水,脂質異常症,痛風,乳酸アシドーシス,肥満 | 体脂肪の再分布/蓄積(胸部,体幹部の脂肪増加,末梢部の脂肪減少,野牛肩) | |
| 血管障害 | 高血圧,蒼白 | ||
| 全身障害及び投与局所様態 | 疲労(1.4%) | 無力症,胸痛,発熱,倦怠感,歩行障害,異形成,全身浮腫,熱過敏,感染,末梢性浮腫,疼痛 | |
| 免疫系障害 | アレルギー反応 | ||
| 肝胆道系障害 | 黄疸(4.2%) | 肝脾腫大,無胆汁症,肝腫大,肝臓細胞障害,脂肪肝 | 胆石症,胆嚢炎,胆汁うっ滞 |
| 生殖系及び乳房障害 | 女性化乳房,男性生殖能低下,無月経,インポテンス,月経障害,骨盤痛 | ||
| 精神障害 | 不眠症,不安,うつ病,睡眠障害,異常な夢,失見当識,激越,リビドー減退,情動不安定,幻覚,敵意,神経過敏,精神病,自殺企図 | ||
| 臨床検査注1) | 総ビリルビン上昇(37.2%),ALT上昇(4.6%),AST上 昇(3.3 %),CK上昇(7.4%),アミラーゼ上昇(11.6%),リパーゼ上昇(2.6%),好中球減少(4.7%),ヘモグロビン減少(1.5%) |
注1):グレード3-4の臨床検査値異常(副作用として報告されたかどうかにかかわらず,臨床試験において測定された臨床検査値異常)
13. 過量投与
13.1 症状
本剤29.2gを過量に服用したHIV感染患者において,無症候性の二束ブロック及びPR間隔の延長が報告されている。
13.2 処置
本剤は,主に肝臓で代謝され,蛋白結合率が高いため,透析は薬剤の除去に有効とは考えられない。
15. その他の注意
15.2 非臨床試験に基づく情報
15.2.1 がん原性,変異原性,生殖毒性
マウス及びラットにおけるがん原性試験において,雌マウスの高用量で良性肝細胞腺腫の発生率が上昇したが,ラットではいかなるタイプの腫瘍の発生率にも上昇はみられなかった。
雌雄マウスで腫瘍発生率の上昇がみられなかった用量における曝露量は,ヒトに400mg/日を投与した場合の曝露量の約4倍である。高用量群の雌マウスでみられた良性肝細胞腺腫の発生率上昇は,肝臓の細胞毒性的な変化(単細胞壊死)に対する二次的な肝細胞増殖の亢進によるものと考えられ,ヒトの臨床治療量における曝露量との関連性は低いと考えられる。
本剤は,ヒト末梢血リンパ球におけるin vitroの染色体異常試験では代謝活性化の有無にかかわらず陽性であった。Ames試験,ラットにおける小核試験及び不定期DNA合成試験,十二指腸のDNA障害試験(コメットアッセイ)の結果は陰性であった。臨床用量(400mg/日)と同程度(雄ラット)又は2倍(雌ラット)の曝露量で,本剤は,交配,受胎能及び初期胚発生に影響しなかった。
15.2.2 動物における毒性・安全性薬理
マウス,ラット及びイヌで実施した反復投与毒性試験において,本剤投与に関連した肝臓の所見として,血清ビリルビン及び肝酵素の増加,肝細胞の空胞化及び肥大がみられ,雌マウスで肝細胞の単細胞壊死が認められた。肝臓の変化がみられた用量でのマウス,ラット及びイヌにおける本剤の全身曝露量は,ヒトに400mgを1日1回投与した場合の曝露量のそれぞれ0.4~12倍,0.4~4倍及び0.2~7倍であった。雌マウスで単細胞壊死がみられた用量での本剤の曝露量は,ヒトに400mgを1日1回投与した場合の曝露量の12倍であった。ラット及びイヌでは血清コレステロール及びグルコースの増加がみられたが,マウスではこれらの変化は認められなかった。
In vitro眼粘膜刺激性試験で,本剤はウシ角膜の混濁度を上昇させたことから,眼に直接接触した場合眼粘膜刺激性を示す可能性がある。
In vitro安全性薬理試験において,本剤はウサギ・プルキンエ線維の活動電位持続時間に対して弱い延長作用を示し,ナトリウムチャネル電流並びに急速活性化遅延整流カリウム電流(HERGによりエンコードされる)及び緩徐活性化遅延整流カリウム電流を軽度(IC50>30μM)に,カルシウム電流を中等度(IC50=10.4μM)に阻害した。イヌにおける心電図の変化(洞性徐脈,PR間隔延長,QT間隔延長及びQRS群延長)が最初に実施した2週間経口投与毒性試験で観察された。別途実施したイヌにおける2週間経口投与毒性試験及び9ヵ月間経口投与毒性試験では薬剤に関連した心電図の変化はみられなかった。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人及びHIV感染患者に対し,アタザナビル400mg又はアタザナビル300mgとリトナビル100mgを,それぞれ1日1回投与したときの薬物動態パラメータを表1に示す。(外国人における成績)
| 薬物動態 |
アタザナビル400mg/日 | アタザナビル300mg/日 +リトナビル100mg/日 |
||
|---|---|---|---|---|
| 健康成人 (n=14) |
HIV感染患者 (n=13) |
健康成人 (n=28) |
HIV感染患者 (n=10) |
|
| Cmax (ng/mL) 幾何平均値 (変動係数%) |
5199 (26) |
2298 (71) |
6129 (31) |
4422 (58) |
| Tmax (h) 中央値 |
2.5 | 2.0 | 2.7 | 3.0 |
| AUC(ng・h/mL) 幾何平均値 (変動係数%) |
28132 (28) |
14874 (91) |
57039 (37) |
46073 (66) |
| 半減期(t1/2) (h) 算術平均値 (標準偏差) |
7.9 (2.9) |
6.5 (2.6) |
18.1 (6.2)a |
8.6 (2.3) |
| Cmin (ng/mL) 幾何平均値 (変動係数%) |
159 (88) |
120 (109) |
1227 (53) |
636 (97) |
a:n=26
HIV感染患者に対して,アタザナビル400mg(200mgカプセル2カプセル)又はアタザナビル300mg(150mgカプセル2カプセル)とリトナビル100mgを,1日1回,軽食とともに投与したときの定常状態におけるアタザナビルの平均血漿中濃度を図1に示す。(外国人における成績)
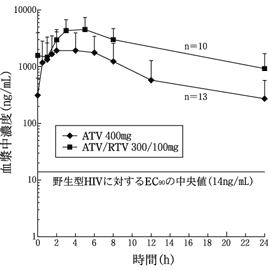
16.1.2 反復投与
アタザナビルは非線形の薬物動態を示し,投与量200〜800mgの範囲でAUC及びCmaxは投与量に比例する用量比以上の増加を示した。投与開始後4〜8日で定常状態に達し,累積係数は約2.3であった。1日400mgを軽食とともに反復投与したとき,健康成人(n=214)及び成人HIV感染患者(n=13)における定常状態の消失半減期は約7時間であった。(外国人における成績)
日本人健康成人男子(12例)にアタザナビル400mg(200mgカプセル2カプセル)を1日1回6日間食事とともに反復投与したときの定常状態(6日目)の薬物動態パラメータ及び平均血漿中濃度推移を表2及び図2に示す。
| パラメータ | 健康成人 |
|---|---|
| Cmax(ng/mL) 幾何平均値(変動係数%) |
6238 (7) |
| Tmax(h) 中央値 |
1.8 |
| AUC(ng・h/mL) 幾何平均値(変動係数%) |
38814 (15) |
| 半減期(t1/2)(h) 算術平均値(標準偏差) |
5.7 (1.7) |
| Cmin(ng/mL) 幾何平均値(変動係数%) |
245 (44) |
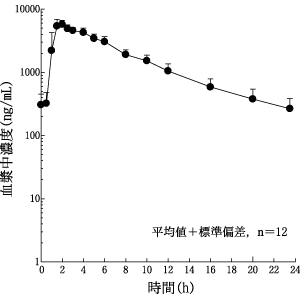
16.2 吸収
16.2.1 食事の影響
本剤又は本剤とリトナビルを食事とともに投与すると,アタザナビルのバイオアベイラビリティーが増大し,薬物動態の変動が減少する。(外国人における成績)
| パラメータ | 投与量a | 幾何平均値 (変動係数%) |
幾何平均値比 (90%信頼区間) |
|||
|---|---|---|---|---|---|---|
| 絶食 | 軽食 | 高脂肪食b | 軽食/絶食 | 高脂肪食/絶食 | ||
| Cmax (ng/mL) |
ATV400mg | 1795 (66) |
2824 (29) |
1795 (33) |
1.57 (1.28-1.93) |
1.00 (0.82-1.23) |
| ATV300mg +RTV100mg |
2391 (49) |
3341 (37) |
2120 (35) |
1.40 (1.17-1.66) |
0.89 (0.75-1.06) |
|
| AUC(INF) (ng・h/mL) |
ATV400mg | 7392 (69) |
12562 (37) |
10000 (43) |
1.70 (1.41-2.05) |
1.35 (1.12-1.63) |
| ATV300mg +RTV100mg |
22255 (45) |
29807 (37) |
22430 (35) |
1.33 (1.17-1.52) |
1.01 (0.89-1.15) |
|
| Cmin (ng/mL) |
ATV300mg +RTV100mg |
275 (50) |
388 (46) |
358 (45) |
1.40 (1.24-1.57) |
1.33 (1.18-1.49) |
a:アタザナビル400mgでは18例,アタザナビル300mg+リトナビル100mgでは40例
b:アタザナビル300mg+リトナビル100mgでは42例
16.3 分布
アタザナビルのヒト血清蛋白への結合は濃度によらず86%であった。アタザナビルはα1-酸性糖蛋白(AAG)及びアルブミンに結合し,両者への結合率はそれぞれ89%及び86%と同程度であった。HIV感染患者に軽食とともに400mgの本剤を1日1回,12週間反復投与した試験では,脳脊髄液及び精液からアタザナビルが検出された。脳脊髄液/血漿の濃度比(n=4)は0.0021~0.0226の範囲で,精液/血漿の濃度比(n=5)は0.11~4.42であった。(外国人における成績)
16.4 代謝
アタザナビルのヒトにおける主な代謝は一酸化及び二酸化反応である。その他,代謝経路の寄与としては大きなものではないが,アタザナビルあるいはその代謝物について,グルクロン酸抱合,N-脱アルキル化,加水分解及び脱水素を伴う酸化反応の代謝経路も存在した。血漿中からは2種の代謝物が検出されたが,いずれもin vitroにおいて抗ウイルス活性を示さなかった。ヒト肝ミクロソームを用いたin vitro試験からアタザナビルはCYP3A4による代謝を受けることが示された。(外国人における成績)
16.5 排泄
14C-アタザナビル400mgを単回投与したとき,標識放射能の79%が糞便中に,13%が尿中に排泄された。また,糞便中及び尿中への未変化体の排泄率はそれぞれ投与量の約20%及び7%であった。(外国人における成績)
16.6 特定の背景を有する患者
16.6.1 腎障害
透析を施行していない重度の腎障害者(30mL/min未満)に本剤400mgを反復投与したときのCmaxは腎機能正常者よりも9%低く,AUC及びCminはそれぞれ19%及び96%高かったが,透析を施行している重度の腎障害者に透析を施行しなかったとき並びに投与2時間後に透析を施行したとき,Cmax及びAUCは腎機能正常者よりも約30〜50%低かった。透析を施行していない腎機能障害患者に用量調節の必要はない。(外国人における成績)[7.5,9.2.1参照]
16.6.2 肝障害
アタザナビルは主に肝臓で代謝を受けて消失する。中等度~重度の肝障害成人被験者(Child-Pugh B群14例及びC群2例)において400mg単回投与後の薬物動態を検討した結果,肝障害者のAUCは健康成人に比べて45%高かった。また,健康成人の半減期が6.4時間であるのに対し,肝障害者では12.1時間であった。(外国人における成績)[7.4,9.3.1,9.3.3参照]
16.6.3 高齢者
若年者(29例,18~40歳)と高齢者(30例,65歳以上)の健康成人において,単回投与時のCmax及びAUCは高齢者の方が17%高かった。両者に著しい違いはなく,薬物動態を基にした年齢による投与量の調整は推奨されない。(外国人における成績)[9.8参照]
16.6.4 小児
6~18歳のHIV感染患者にアタザナビルとリトナビルをそれぞれ体表面積換算で1日1回投与したときの定常状態におけるアタザナビルの薬物動態パラメータを表4に示す。(外国人における成績)
| アタザナビル205mg/m2 リトナビル100mg/m2 |
||
|---|---|---|
| 6-13歳未満 (n=17) |
13-18歳未満 (n=10) |
|
| 投与量(mg) [min-max] |
200mg [150-400] |
400mg [250-500] |
| Cmax (ng/mL) 幾何平均値 (変動係数%) |
4451 (33%) |
3711 (46%) |
| AUC (ng・h/mL) 幾何平均値 (変動係数%) |
42503 (36%) |
44970 (34%) |
| Cmin (ng/mL) 幾何平均値 (変動係数%) |
535 (62%) |
1090 (60%) |
16.7 薬物相互作用
アタザナビルは肝臓でCYP3A4により代謝される。アタザナビルはCYP3A4を不可逆的に阻害する。そのCYP3A4に対する阻害定数(Ki)は0.84〜1.0μMである。また,UGT1A1を阻害し,そのKi値は1.9μMである。
アタザナビルはCYP2C8の阻害剤であり,Ki値は2.1μMである。アタザナビルはCYP1A2及びCYP2C9を競合的に阻害し,Ki値は12μM,Cmax/Ki値比は約0.25である。本剤はCYP1A2あるいはCYP2C9により代謝される薬物と薬物相互作用を発現する可能性が考えられる。臨床用量で得られる濃度でアタザナビルはCYP2C19あるいはCYP2E1を阻害しない。In vivoにおいて,アタザナビルは本剤自身の代謝を誘導せず,またCYP3A4で代謝される薬剤の代謝を促進しない。反復投与試験において,本剤は尿中の内因性6β-ヒドロキシコルチゾール/コルチゾール比を低下させ,CYP3A4を誘導しないことが示唆された。
CYP3A4活性を誘導する薬剤はアタザナビルのクリアランスを上昇させ,血漿中濃度を低下させる可能性がある。また,本剤とCYP3A4を阻害する他剤との併用投与によりアタザナビルの血漿中濃度が上昇する可能性がある。
本剤と併用の可能性のある他剤又は薬物動態学的相互作用の指標として一般に使用されている薬剤との薬物相互作用試験を実施した。併用投与がCmax,AUC及びCminに及ぼす影響を表5及び表6に示す。(外国人における成績)[10.,10.1,10.2参照]
| 併用薬 | 併用薬の投与量 /スケジュールa |
本剤の投与量/ スケジュールa |
n | アタザナビルの 併用時/非併用時 |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| アテノロール | 50mg(QD) 7〜11日目, 19〜23日目 |
400mg(QD) 1〜11日目 |
19 | 1.00 (0.89,1.12) |
0.93 (0.85,1.01) |
0.74 (0.65,0.86) |
| クラリスロマイシン | 500mg(BID) 7〜10日目, 18〜21日目 |
400mg(QD) 1〜10日目 |
29 | 1.06 (0.93,1.20) |
1.28 (1.16,1.43) |
1.91 (1.66,2.21) |
| ジルチアゼム | 180mg(QD) 7〜11日目, 19〜23日目 |
400mg(QD) 1〜11日目 |
30 | 1.04 (0.96,1.11) |
1.00 (0.95,1.05) |
0.98 (0.90,1.07) |
| エファビレンツ(EFV) | 600mg(QD) 7〜20日目 |
400mg(QD) 1〜20日目 |
27 | 0.41 (0.33,0.51) |
0.26 (0.22,0.32) |
0.07 (0.05,0.10) |
| 600mg(QD) 7〜20日目 |
400mg(QD) 1〜6日目 300mg(QD)+ RTV100mg(QD) (EFV投与の2時間前) 7〜20日目 |
13 | 1.14 (0.83,1.58) |
1.39 (1.02,1.88) |
1.48 (1.24,1.76) |
|
| 600mg(QD) 11〜24日目 |
300mg(QD) + RTV100mg (QD) 1〜10日目 400mg(QD)+ RTV100mg(QD) 11〜24日目 (EFVと同時投与) |
14 | 1.17 (1.08,1.27) |
1.00 (0.91,1.10) |
0.58 (0.49,0.69) |
|
| ファモチジン | 40mg(BID) 7〜12日目 |
400mg(QD) 1〜12日目 (ファモチジンと同時投与) |
15 | 0.53 (0.34,0.82) |
0.59 (0.40,0.87) |
0.58 (0.37,0.89) |
| 40mg(BID) 7〜12日目 |
400mg(QD, pm) 1〜6日目 7〜12日目 (ファモチジン投与の 10時間後かつ2時間前) |
14 | 1.08 (0.82,1.41) |
0.95 (0.74,1.21) |
0.79 (0.60,1.04) |
|
| 40mg(BID) 11〜20日目 |
300mg(QD)+ RTV100mg(QD) 1〜20日目 (ファモチジンと同時投与) |
14 | 0.86 (0.79,0.94) |
0.82 (0.75,0.89) |
0.72 (0.64,0.81) |
|
| 20mg(BID) 11〜17日目 |
300mg(QD)+ RTV100mg(QD)+ テノホビル300mg(QD, am) 1〜17日目 (ファモチジンと同時投与) |
18 | 0.91 (0.84,0.99) |
0.90 (0.82,0.98) |
0.81 (0.69,0.94) |
|
| 40mg(QD, pm) 18〜24日目 |
300mg(QD)+ RTV100mg(QD)+ テノホビル300mg(QD, am) 1〜10日目 18〜24日目 (ファモチジン投与の12時間後) |
20 | 0.89 (0.81,0.97) |
0.88 (0.80,0.96) |
0.77 (0.63,0.93) |
|
| 40mg(BID) 18〜24日目 |
300mg(QD)+ RTV100mg(QD)+ テノホビル300mg(QD, am) 1〜10日目 18〜24日目 (ファモチジン投与の10時間後 かつファモチジン投与の2時間前) |
18 | 0.74 (0.66,0.84) |
0.79 (0.70,0.88) |
0.72 (0.63,0.83) |
|
| 40mg(BID) 11〜20日目 |
300mg(QD)+ RTV100mg(QD, am) 1〜10日目 400mg(QD)+ RTV100mg(QD, am) 11〜20日目 |
15 | 1.02 (0.87,1.18) |
1.03 (0.86,1.22) |
0.86 (0.68,1.08) |
|
| フルコナゾール | 200mg(QD) 11〜20日目 |
300mg(QD)+ RTV100mg(QD) 1〜20日目 |
29 | 1.03 (0.95,1.11) |
1.04 (0.95,1.13) |
0.98 (0.85,1.13) |
| ケトコナゾール | 200mg(QD) 7〜13日目 |
400mg(QD) 1〜13日目 |
14 | 0.99 (0.77,1.28) |
1.10 (0.89,1.37) |
1.03 (0.53,2.01) |
| ネビラピン | 200mg(BID) 1〜23日目 |
300mg(QD)+ RTV100mg(QD) 4〜13日目 |
23 | 0.72 (0.60,0.86) |
0.58 (0.48,0.71) |
0.28 (0.20,0.40) |
| 400mg(QD)+ RTV100mg(QD) 14〜23日目 |
23 | 1.02 (0.85,1.24) |
0.81 (0.65,1.02) |
0.41 (0.27,0.60) |
||
| オメプラゾール | 40mg(QD)b 7〜12日目 |
400mg(QD) 1〜12日目 |
16 | 0.04 (0.04,0.05) |
0.06 (0.05,0.07) |
0.05 (0.03,0.07) |
| 40mg(QD)b 11〜20日目 |
300mg(QD)+ RTV100mg(QD) 1〜20日目 |
15 | 0.28 (0.24,0.32) |
0.24 (0.21,0.27) |
0.22 (0.19,0.26) |
|
| 20mg(QD, am) 17〜23日目 |
300mg(QD)+ RTV100mg(QD, pm) 7〜23日目 |
13 | 0.61 (0.46,0.81) |
0.58 (0.44,0.75) |
0.54 (0.41,0.71) |
|
| 20mg(QD, am) 17〜23日目 |
300mg(QD)+ RTV100mg(QD, am) 7〜16日目 400mg(QD)+ RTV100mg(QD, am) 17〜23日目 |
14 | 0.69 (0.58,0.83) |
0.70 (0.57,0.86) |
0.69 (0.54,0.88) |
|
| リファブチン | 150mg(QD) 15〜28日目 |
400mg(QD) 1〜28日目 |
7 | 1.34 (1.14,1.59) |
1.15 (0.98,1.34) |
1.13 (0.68,1.87) |
| リファンピシン | 600mg(QD) 17〜26日目 |
300mg(QD)+ RTV100mg(QD) 7〜26日目 |
16 | 0.47 (0.41,0.53) |
0.28 (0.25,0.32) |
0.02 (0.02,0.03) |
| リトナビル(RTV)c | 100mg(QD) 11〜20日目 |
300mg(QD) 1〜20日目 |
28 | 1.86 (1.69,2.05) |
3.38 (3.13,3.63) |
11.89 (10.23,13.82) |
| テノホビル ジソプロキシルフマル酸塩 | 300mg(QD) 9〜16日目 |
400mg(QD) 2〜16日目 |
34 | 0.79 (0.73,0.86) |
0.75 (0.70,0.81) |
0.60 (0.52,0.68) |
| 300mg(QD) 15〜42日目 |
300mg(QD)+ RTV100mg(QD) 1〜42日目 |
10 | 0.72d (0.50,1.05) |
0.75d (0.58,0.97) |
0.77d (0.54,1.10) |
|
| ボリコナゾール (CYP2C19の活性型遺伝子を 1つ以上有する被験者) |
200mg(BID) 2〜3日目, 22〜30日目 400mg(BID) 1日目,21日目 |
300mg(QD)+ RTV100mg(QD) 11〜30日目 |
20 | 0.87 (0.80,0.96) |
0.88 (0.82,0.95) |
0.80 (0.72,0.90) |
| ボリコナゾール (CYP2C19の活性型遺伝子を 有さない被験者) |
50mg(BID) 2〜3日目, 22〜30日目 100mg(BID) 1日目,21日目 |
300mg(QD)+ RTV100mg(QD) 11〜30日目 |
8 | 0.81 (0.66,1.00) |
0.80 (0.65,0.97) |
0.69 (0.54,0.87) |
a:特に明記されていない場合は食事と共に投与を実施した。
b:本剤を投与する2時間前の空腹時にオメプラゾールを投与。
c:アタザナビル400mg(QD)のこれまでの成績と比較して,アタザナビル/リトナビルの300mg/100mg投与時(QD)のCmax,AUC及びCminの幾何平均値はそれぞれ18%,103%及び671%増加した。リトナビルと併用投与したときのCmax,AUC及びCminの幾何平均値はそれぞれ6129ng/mL,57039ng・h/mL及び1227ng/mLであった。
d:リトナビル/テノホビル併用時とリトナビル併用時のアタザナビル各パラメータの比率。アタザナビル/リトナビル300mg/100mg投与時のアタザナビルの曝露量は,アタザナビル400mg投与時よりも高かった(注c参照)。3剤併用時のCmax,AUC及びCminの幾何平均値はそれぞれ3190ng/mL,34459ng・h/mL及び491ng/mLであった。
| 併用薬 | 併用薬の投与量/ スケジュールa |
本剤の投与量/ スケジュールa |
n | 併用薬の薬物動態パラメータ比 併用時/非併用時(90%信頼区間) |
||
|---|---|---|---|---|---|---|
| Cmax | AUC | Cmin | ||||
| アセトアミノフェン | 1g(BID) 1〜20日目 |
300mg(QD)+ RTV100mg(QD) 11〜20日目 |
10 | 0.87 (0.77,0.99) |
0.97 (0.91,1.03) |
1.26 (1.08,1.46) |
| アテノロール | 50mg(QD) 7〜11日目, 19〜23日目 |
400mg(QD) 1〜11日目 |
19 | 1.34 (1.26,1.42) |
1.25 (1.16,1.34) |
1.02 (0.88,1.19) |
| クラリスロマイシン | 500mg(BID) 7〜10日目, 18〜21日目 |
400mg(QD) 1〜10日目 |
21 | 1.50 (1.32,1.71) 水酸化体: 0.28 (0.24,0.33) |
1.94 (1.75,2.16) 水酸化体: 0.30 (0.26,0.34) |
2.60 (2.35,2.88) 水酸化体: 0.38 (0.34,0.42) |
| ジルチアゼム | 180mg(QD) 7〜11日目, 19〜23日目 |
400mg(QD) 1〜11日目 |
28 | 1.98 (1.78,2.19) デスアセチルジルチアゼム: 2.72 (2.44,3.03) |
2.25 (2.09,2.16) デスアセチルジルチアゼム: 2.65 (2.45,2.87) |
2.42 (2.14,2.73) デスアセチルジルチアゼム: 2.21 (2.02,2.42) |
| エチニルエストラジオール ・ノルエチステロン |
Ortho-Novum 7/7/7(QD) 1〜29日目 |
400mg(QD) 16〜29日目 |
19 | エチニルエストラジオール: 1.15 (0.99,1.32) |
エチニルエストラジオール: 1.48 (1.31,1.68) |
エチニルエストラジオール: 1.91 (1.57,2.33) |
| ノルエチステロン: 1.67 (1.42,1.96) |
ノルエチステロン: 2.10 (1.68,2.62) |
ノルエチステロン: 3.62 (2.57,5.09) |
||||
| エチニルエストラジオール ・ノルゲスチメート |
Ortho Tri-Cyclen(QD) 1〜28日目 Ortho Tri-Cyclen LO(QD) 29〜42日目 |
300mg(QD)+ RTV100mg(QD) 29〜42日目 |
14 | エチニルエストラジオール: 0.84 (0.74,0.95) |
エチニルエストラジオール: 0.81 (0.75,0.87) |
エチニルエストラジオール: 0.63 (0.55,0.71) |
| 17-デアセチルノルゲスチメート: 1.68 (1.51,1.88) |
17-デアセチルノルゲスチメート: 1.85 (1.67,2.05) |
17-デアセチルノルゲスチメート: 2.02 (1.77,2.31) |
||||
| フルコナゾール | 200mg(QD) 1〜20日目 |
300mg(QD)+ RTV100mg(QD) 11〜20日目 |
29 | 1.05 (0.99,1.10) |
1.08 (1.02,1.15) |
1.07 (1.00,1.15) |
| ネビラピン | 200mg(BID) 1〜23日目 |
300mg(QD)+ RTV100mg(QD) 4〜13日目 |
23 | 1.17 (1.09,1.25) |
1.25 (1.17,1.34) |
1.32 (1.22,1.43) |
| 400mg(QD)+ RTV100mg(QD) 14〜23日目 |
1.21 (1.11,1.32) |
1.26 (1.17,1.36) |
1.35 (1.25,1.47) |
|||
| オメプラゾール | 40mg単回 7日目,20日目b |
400mg(QD) 1〜12日目 |
16 | 1.24 (1.04,1.47) |
1.45 (1.20,1.76) |
NA |
| リファブチン | 300mg(QD) 1〜10日目 150mg(QD) 11〜20日目 |
600mg(QD)c 11〜20日目 |
3 | 1.18 (0.94,1.48) 25-O-デスアセチルリファブチン: 8.20 (5.90,11.40) |
2.10 (1.57,2.79) 25-O-デスアセチルリファブチン: 22.01 (15.97,30.34) |
3.43 (1.98,5.96) 25-O-デスアセチルリファブチン: 75.6 (30.1,190.0) |
| 150mg(週2回) 1〜15日目 |
300mg(QD)+ RTV100mg(QD) 1〜17日目 |
7 | 2.49 (2.03,3.06) 25-O-デスアセチルリファブチン: 7.77 (6.13,9.83) |
1.48 (1.19,1.84) 25-O-デスアセチルリファブチン: 10.90 (8.14,14.61) |
1.40 (1.05,1.87) 25-O-デスアセチルリファブチン: 11.45 (8.15,16.10) |
|
| Rosiglitazone(本邦未承認) | 4mg単回 1日目,7日目,17日目 |
400mg(QD) 2〜7日目 |
14 | 1.08 (1.03,1.13) |
1.35 (1.26,1.44) |
NA |
| 300mg(QD)+ RTV100mg(QD) 8〜17日目 |
0.97 (0.91,1.04) |
0.83 (0.77,0.89) |
NA | |||
| テノホビル ジソプロキシルフマル酸塩 | 300mg(QD) 9〜16日目,24〜30日目 |
400mg(QD) 2〜16日目 |
33 | 1.14 (1.08,1.20) |
1.24 (1.21,1.28) |
1.22 (1.15,1.30) |
| 300mg(QD,pm) 1〜7日目,25〜34日目 |
300mg(QD)+ RTV100mg(QD) 25〜34日目 |
12 | 1.34 (1.20,1.51) |
1.37 (1.30,1.45) |
1.29 (1.21,1.36) |
|
| ボリコナゾール (CYP2C19の活性型遺伝子を 1つ以上有する被験者) |
200mg(BID) 2〜3日目,22〜30日目 400mg(BID) 1日目,21日目 |
300mg(QD)+ RTV100mg(QD) 11〜30日目 |
20 | 0.90 (0.78,1.04) |
0.67 (0.58,0.78) |
0.61 (0.51,0.72) |
| ボリコナゾール (CYP2C19の活性型遺伝子を 有さない被験者) |
50mg(BID) 2〜3日目,22〜30日目 100mg(BID) 1日目,21日目 |
300mg(QD)+ RTV100mg(QD) 11〜30日目 |
8 | 4.38 (3.55,5.39) |
5.61 (4.51,6.99) |
7.65 (5.71,10.2) |
| ラミブジン +ジドブジン |
ラミブジン150mg+ ジドブジン300mg(BID), 1〜12日目 |
400mg(QD) 7〜12日目 |
19 | ラミブジン: 1.04 (0.92,1.16) |
ラミブジン: 1.03 (0.98,1.08) |
ラミブジン: 1.12 (1.04,1.21) |
| ジドブジン: 1.05 (0.88,1.24) ジドブジングルクロナイド: 0.95 (0.88,1.02) |
ジドブジン: 1.05 (0.96,1.14) ジドブジングルクロナイド: 1.00 (0.97,1.03) |
ジドブジン: 0.69 (0.57,0.84) ジドブジングルクロナイド: 0.82 (0.62,1.08) |
||||
a:特に明記されていない場合は食事と共に投与を実施した。
b:7日目において本剤投与2時間後にオメプラゾールを投与し,20日目には軽食摂取2時間後に投与した。
c:承認用量ではない。
NA:該当データなし。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 抗HIV薬による治療経験のない患者における海外第III相臨床試験(AI424-138試験)
抗HIV薬による治療経験のないHIV-1感染患者883例を対象に,本剤(300mg1日1回)+リトナビル(100mg1日1回)投与群(ATV/RTV群)及びロピナビル+リトナビル(400/100mg1日2回)投与群(LPV/RTV群)として,それぞれテノホビルジソプロキシルフマル酸塩/エムトリシタビン(300/200mg1日1回)との併用による,無作為化オープン比較試験を実施した。年齢平均値は36歳(範囲:19〜72歳),48%が白人,69%が男性であった。投与前の平均血漿中CD4リンパ球数は214cells/mm3(範囲:2〜810cells/mm3),HIV-1 RNAレベルは4.94log10 copies/mL(範囲:2.60〜5.88log10 copies/mL)であった1)。
| ATV/ (n=440) |
LPV/ (n=443) |
||||
|---|---|---|---|---|---|
| 48週 | 96週 | 48週 | 96週 | ||
| HIV-1 RNA量が 400copies/mL未満 50copies/mL未満 に抑制されていた患者の割合a |
86% 78% |
80% 74% |
82% 76% |
74% 68% |
|
| HIV-1 RNA量の投与前値からの 平均変化量(log10 copies/mL) |
−3.09 | −3.21 | −3.13 | −3.19 | |
| CD4リンパ球数の投与前値からの 平均変化量(cells/mm3) |
203 | 268 | 219 | 290 | |
a:Roche Amplicor, v 1.5 ultra-sensitive assay使用
| ATV/RTV群a | LPV/RTV群a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 投与前 | 48週 | 96週 | 投与前 | 48週 | 96週 | |||||
| mg/dLb | mg/dLb | 変化c | mg/dLb | 変化c | mg/dLb | mg/dLb | 変化c | mg/dLb | 変化c | |
| (n=429d) | (n=374d) | (n=374d) | (n=342d) | (n=342d) | (n=424d) | (n=335d) | (n=335d) | (n=291d) | (n=291d) | |
| LDLコレステロールe | 92 (1.5) |
105 (1.7) |
+14% (12.1%,15.7%) |
105 (1.7) |
+14% (12.4%,16.3%) |
93 (1.4) |
111 (1.9) |
+19% (16.8%,20.6%) |
110 (2.0) |
+17% (15.3%,18.9%) |
| HDLコレステロールe | 37 (0.6) |
46 (0.6) |
+29% (26.9%,31.1%) |
44 (0.6) |
+21% (19.1%,23.4%) |
36 (0.6) |
48 (0.7) |
+37% (34.6%,39.2%) |
46 (0.8) |
+29% (26.8%,31.6%) |
| 総コレステロールe | 149 (1.8) |
169 (2.0) |
+13% (12.1%,14.4%) |
169 (2.0) |
+13% (12.0%,14.6%) |
150 (1.7) |
187 (2.3) |
+25% (24.0%,26.6%) |
186 (2.4) |
+25% (23.6%,26.3%) |
| トリグリセライドe | 126 (3.7) |
145 (4.3) |
+15% (12.2%,17.8%) |
140 (4.1) |
+13% (10.4%,16.1%) |
129 (3.9) |
194 (6.4) |
+52% (48.3%,56.0%) |
184 (5.9) |
+50% (46.0%,54.2%) |
a:高脂血症薬による治療を開始した症例(投与前ATV/RTV群:1%,LPV/RTV群:1%,48週においてATV/RTV群:2%,LPV/RTV群:8%,96週においてATV/RTV群:3%,LPV/RTV群:10%)は除いて集計した。
b:平均値(標準誤差)
c:投与前値と48週又は96週の両値を計測した患者における変化の平均
d:LDLコレステロール値を計測した患者数
e:空腹時
副作用発現頻度(中等度又は高度)は,本剤300mg/リトナビル100mgと他の抗HIV薬併用投与群で30.2%(133/441例)であった。主な副作用は,悪心4.1%(18/441例),黄疸・黄疸眼5.0%(22/441例),下痢2.5%(11/441例),発疹2.7%(12/441例)等であった。
また,グレード3-4の臨床検査値異常の主なものは,AST上昇2.5%(11/435例),ALT上昇2.5%(11/435例),総ビリルビン上昇44.1%(192/435例),リパーゼ上昇2.1%(9/435例),CK上昇7.8%(34/435例),総コレステロール上昇10.8%(47/434例),好中球減少4.8%(21/434例)等であった。
17.1.2 抗HIV薬による治療経験のない患者における海外第III相臨床試験(AI424-034試験)
抗HIV薬による治療経験のないHIV-1感染患者810例を対象に,本剤(400mg1日1回)+ラミブジン(150mg)及びジドブジン(300mg)1日2回投与群(ATV群)とエファビレンツ(600mg1日1回)+ラミブジン(150mg)及びジドブジン(300mg)の1日2回投与群(EFV群)として,無作為化二重盲検比較試験を実施した。投与された805例の年齢平均値は34歳(範囲:18〜73歳),33%が白人,65%が男性であった。投与前の平均CD4リンパ球数は322cells/mm3(範囲:64〜1424cells/mm3),投与前の平均血漿中HIV-1 RNAレベルは4.84log10 copies/mL(範囲:2.23〜5.88log10 copies/mL)であった2)。
| ATV群 (n=404) |
EFV群 (n=401) |
|
|---|---|---|
| HIV-1 RNA量が 400copies/mL未満 50copies/mL未満 に48週時点において抑制されていた患者の割合a |
67% 31% |
63% 36% |
| HIV-1 RNA量の投与前値からの平均変化量 (log10 copies/mL) |
−2.67 | −2.74 |
| CD4リンパ球数の投与前値からの平均変化量 (cells/mm3) |
176 | 160 |
a:Roche Amplicor HIV-1 Monitor Assay, test version 1.0又は1.5使用
| ATV群a | EFV群a | |||||
|---|---|---|---|---|---|---|
| 投与前 | 48週 | 投与前 | 48週 | |||
| mg/dL | mg/dL | 変化b | mg/dL | mg/dL | 変化b | |
| (n=383c) | (n=283c) | (n=272c) | (n=378c) | (n=264c) | (n=253c) | |
| LDLコレステロールd | 98 | 98 | +1% | 98 | 114 | +18% |
| HDLコレステロール | 39 | 43 | +13% | 38 | 46 | +24% |
| 総コレステロール | 164 | 168 | +2% | 162 | 195 | +21% |
| トリグリセライドd | 138 | 124 | −9% | 129 | 168 | +23% |
a:高脂血症薬による治療を開始した症例(投与前ATV群:<1%,EFV群:0%,48週においてATV群:1%,EFV群:3%)は除いて集計した。
b:投与前値と48週の両値を計測した患者の変化の平均
c:LDLコレステロール値を計測した患者数
d:空腹時
副作用発現頻度(中等度又は高度)は,本剤と他の抗HIV薬併用投与群で40.8%(165/404例)であった。主な副作用は,悪心14.1%(57/404例),頭痛5.7%(23/404例),発疹6.2%(25/404例),腹痛4.0%(16/404例),黄疸・黄疸眼6.7%(27/404例),嘔吐4.2%(17/404例)等であった。
また,グレード3-4の臨床検査値異常の主なものは,総ビリルビン上昇32.6%(131/402例),CK上昇6.2%(25/401例),ALT上昇3.7%(15/402例),ヘモグロビン減少4.5%(18/402例),好中球減少6.5%(26/402例)等であった。
17.1.3 抗HIV薬による治療経験のある患者における海外第III相臨床試験(AI424-045試験)
ウイルス学的治療失敗を伴う抗HIV薬による治療経験のあるHIV-1感染患者358例を対象に,本剤(300mg1日1回)+リトナビル(100mg1日1回)(ATV/RTV群),本剤(400mg1日1回)+サキナビル(1200mg1日1回)(ATV/SQV群)及びロピナビル+リトナビル(400mg/100mg1日2回)(LPV/RTV群)として,それぞれテノホビルジソプロキシルフマル酸塩(300mg1日1回)とヌクレオシド系逆転写酵素阻害薬1剤との併用による,無作為化非盲検比較試験を実施した。年齢平均値は41歳(範囲:24〜74歳),60%が白人,78%が男性であった。投与前の平均血漿中CD4リンパ球数は337cells/mm3(範囲:14〜1543cells/mm3),HIV-1 RNAレベルは4.40log10 copies/mL(範囲:2.60〜5.88log10 copies/mL)であった。ATV/RTV群及びLPV/RTV群における48週の成績を表5,脂質の投与前値からの変化を表6に示す。ATV/SQV群(115例)におけるHIV-1 RNA量の投与前値からの平均変化量は−1.55log10 copies/mLであり,LPV/RTV群とのTime-Averaged Differenceは0.33であった。CD4リンパ球数の平均変化量は72cells/mm3増加し,血漿中HIV-1RNAレベルが400copies/mL未満(50copies/mL未満)に抑えられた患者はそれぞれ37%(24%)であった。本試験において,ATV/SQVの併用投与は十分な効果を得ることはできなかった。
また,治療期間96週を通じて,ATV/RTV群とLPV/RTV群のHIV-1 RNA量の投与前値からの平均変化量は観察した症例において非劣性を示した3)。
| ATV/RTV群 (n=120) |
LPV/RTV群 (n=123) |
Time-Averaged Difference (95%信頼区間) |
|
|---|---|---|---|
| HIV-1 RNA量が 400copies/mL未満 50copies/mL未満 に抑制されていた患者の割合b |
53% 36% |
54% 42% |
−1.1 (−13.7, 11.4) −6.4 (−18.7, 5.8) |
| HIV-1 RNA量の投与前値からの平均変化量 (log10 copies/mL)b,c |
−1.93 | −1.87 | 0.13 (−0.12, 0.39)a |
| CD4リンパ球数の投与前値からの平均変化量(cells/mm3)d | 110 | 121 | −17.5 (−45.6, 10.6) |
a:HIV-1 RNA量の平均変化量は97.5%信頼区間
b:Roche Amplicor HIV-1 Monitor Assay, test version 1.5使用
c:プロトコール上のprimary efficacy outcome measure
d:投与前と48週のCD4リンパ球数を計測した患者(ATV/RTV群,n=83;LPV/RTV群,n=94)における平均変化量
| ATV/RTV群a | LPV/RTV群a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 投与前 | 48週 | 96週 | 投与前 | 48週 | 96週 | |||||
| mg/dLb | mg/dLb | 変化c | mg/dLb | 変化c | mg/dLb | mg/dLb | 変化c | mg/dLb | 変化c | |
| (n=111d) | (n=76d) | (n=75d) | (n=52d) | (n=52d) | (n=108d) | (n=76d) | (n=73d) | (n=44d) | (n=43d) | |
| LDLコレステロールe | 108 (4.4) |
97 (4.1) |
−11% (−14.4%,−7.0%) |
100 (4.9) |
−11% (−15.1%,−7.6%) |
105 (4.0) |
103 (3.8) |
+1% (−3.4%,4.6%) |
102 (5.2) |
+1% (−2.5%,5.5%) |
| HDLコレステロール | 40 (1.1) |
40 (1.2) |
−7% (−9.0%,−4.4%) |
42 (1.5) |
−5% (−8.2%,−0.7%) |
39 (1.2) |
41 (1.3) |
+2% (−1.4%,6.0%) |
44 (1.5) |
+7% (3.6%,11.1%) |
| 総コレステロール | 188 (5.0) |
170 (4.1) |
−8% (−9.7%,−6.0%) |
174 (4.9) |
−7% (−9.0%,−4.7%) |
181 (4.3) |
187 (4.2) |
+6% (4.3%,8.6%) |
188 (5.1) |
+9% (6.0%,11.7%) |
| トリグリセライドe | 216 (16.3) |
162 (9.4) |
−3% (−8.2%,2.2%) |
168 (16.2) |
−2% (−8.5%,4.6%) |
196 (11.8) |
224 (14.6) |
+30% (22.3%,38.6%) |
223 (18.0) |
+30% (20.4%,40.6%) |
a:高脂血症薬による治療を開始した症例(投与前ATV/RTV群:4%,LPV/RTV群:4%,48週においてATV/RTV群:8%,LPV/RTV群:19%,96週においてATV/RTV群:9%,LPV/RTV群:20%)は除いて集計した。
b:平均値(標準誤差)
c:投与前値と48週又は96週の両値を計測した患者における変化の平均
d:LDLコレステロール値を計測した患者数
e:空腹時
副作用発現頻度(中等度又は高度)は,本剤300mg/リトナビル100mgと他の抗HIV薬併用投与群で28.6%(34/119例)であった。主な副作用は,発熱1.7%(2/119例),悪心2.5%(3/119例),黄疸・黄疸眼9.2%(11/119例),下痢2.5%(3/119例),うつ病1.7%(2/119例),筋肉痛4.2%(5/119例)等であった。また,グレード3-4の臨床検査値異常の主なものは,AST上昇3.4%(4/119例),ALT上昇4.2%(5/119例),総ビリルビン上昇48.7%(58/119例),リパーゼ上昇5.0%(6/119例),CK上昇8.4%(10/119例),総コレステロール上昇5.0%(6/119例),トリグリセリド上昇7.8%(9/116例),血中ブドウ糖増加5.2%(6/116例),好中球減少6.7%(8/119例),血小板減少1.7%(2/119例)等であった。
17.1.4 抗HIV薬による治療経験のある患者における海外第III相臨床試験(AI424-043試験)
過去に1回のHIVプロテアーゼ阻害薬を1剤含む抗HIV療法において効果不十分なHIV-1感染患者300例を対象に,本剤(400mg1日1回)投与群(ATV群)とロピナビル+リトナビル(400mg/100mg1日2回)投与群(LPV/RTV群)として,それぞれヌクレオシド系逆転写酵素阻害薬2剤との併用による,無作為化オープン比較試験を実施した。治療期間48週において,血漿中HIV-1 RNA量が400copies/mL未満(50copies/mL未満)であった患者の割合は,ATV群(n=150)で45%(32%),LPV/RTV群(n=150)で67%(51%)であり,HIV-1 RNA量の投与前値からの平均変化量はATV群で−1.59log10 copies/mL,LPV/RTV群で−2.02log10 copies/mLであった。ATV群の抗ウイルス効果はLPV/RTV群に比較して有意に低かった。副作用発現頻度(中等度又は高度)は,本剤400mgと他の抗HIV薬併用投与群で23.6%(34/144例)であった。主な副作用は,頭痛4.2%(6/144例),下痢2.1%(3/144例),黄疸3.5%(5/144例),悪心2.8%(4/144例),腹痛2.8%(4/144例),嘔吐2.1%(3/144例),リポジストロフィー5.6%(8/144例),体重減少2.1%(3/144例),末梢神経障害2.1%(3/144例),発疹2.1%(3/144例)等であった。また,グレード3-4の臨床検査値異常の主なものは,AST上昇3.5%(5/143例),ALT上昇7.0%(10/143例),総ビリルビン上昇25.2%(36/143例),好中球減少5.6%(8/143例),CK上昇8.4%(12/143例),リパーゼ上昇4.2%(6/143例),総コレステロール上昇2.8%(4/143例),トリグリセリド上昇5.6%(8/142例)等であった。
17.1.5 海外第I/II相臨床試験(小児)(AI424-020試験)
生後3ヵ月〜21歳のHIV感染患者193例(抗HIV薬による治療経験のない患者86例,抗HIV薬による治療経験のある患者107例)を対象に,本剤(カプセル剤又は散剤)1日1回投与群(ATV群)及び本剤(カプセル剤又は散剤)1日1回+リトナビル投与群(ATV/RTV群)として,それぞれヌクレオシド系逆転写酵素阻害薬2剤との併用によるオープン試験を実施した。
本剤(カプセル剤)の投与を受けた6〜18歳未満のHIV感染患者105例が評価された。
投与前の平均血漿中CD4リンパ球数は,抗HIV薬による治療経験のない患者のATV群で326cells/mm3,ATV/RTV群で355cells/mm3,抗HIV薬による治療経験のある患者のATV群で430cells/mm3,ATV/RTV群で522cells/mm3であった。
| 抗HIV薬による 治療経験のない患者 |
抗HIV薬による 治療経験のある患者 |
|||
|---|---|---|---|---|
| ATV群 | ATV/ RTV群 |
ATV群 | ATV/ RTV群 |
|
| n=26 | n=17 | n=37 | n=25 | |
| HIV-1 RNA量が 400copies/mL未満 50copies/mL未満 に抑制されていた患者の割合a |
38% 38% |
65% 59% |
30% 19% |
40% 32% |
| CD4リンパ球数の投与前値からの平均変化量 (cells/mm3) |
431 (n=16b) |
343 (n=12b) |
201 (n=19b) |
335 (n=12b) |
a:Intent-to-treat analysis
b:投与前と96週のCD4リンパ球数を計測した患者
有害事象に基づく発現頻度(中等度又は高度)は,本剤と他の抗HIV薬併用投与群で92.1%(58/63例)及び本剤/リトナビルと他の抗HIV併用投与群で97.6%(41/42例)であった。主な有害事象は,本剤と他の抗HIV薬併用投与群では血中非抱合ビリルビン増加73.0%(46/63例),血中ビリルビン増加60.3%(38/63例),血中ブドウ糖減少28.6%(18/63例),発熱22.2%(14/63例),咳嗽20.6%(13/63例)等,本剤/リトナビルと他の抗HIV薬併用投与群では血中非抱合ビリルビン増加88.1%(37/42例),血中ビリルビン増加76.2%(32/42例),抱合ビリルビン増加28.6%(12/42例),咳嗽21.4%(9/42例)等であった。
また,グレード3-4の臨床検査値異常の主なものは,本剤と他の抗HIV薬併用投与群では総ビリルビン上昇52.4%(33/63例),好中球減少4.8%(3/63例),低血糖4.8%(3/63例),ALT上昇3.2%(2/63例),AST上昇3.2%(2/63例),GGT上昇3.2%(2/63例)等,本剤/リトナビルと他の抗HIV薬併用投与群では総ビリルビン上昇66.7%(28/42例),好中球減少14.3%(6/42例), GGT上昇2.4% (1/42例),クレアチニン上昇4.8% (2/42 例),高血糖 2.4%(1/42例),低血糖2.4%(1/42例),総コレステロール上昇2.4%(1/42例)等であった。
17.3 その他
17.3.1 心電図への影響
健康成人において,アタザナビルを投与した際に血中濃度及び投与量に依存したPR間隔の延長が観察されている。
プラセボ対照試験(AI424-076)において,PR間隔の投与前値からの最大変化の平均値(±SD)はアタザナビル400mg投与群(n=65)で24(±15)msecで,プラセボ投与群(n=67)で13(±11)msecであった。この試験におけるPR間隔の延長は無症候性であった。
また,アタザナビルの心電図への影響を72例の健康成人を用いた臨床薬理試験において確認した。アタザナビル400mg,800mg注)の経口投与とプラセボ投与を比較したところ,アタザナビルはQTc間隔(Fridericiaの補正を用いた)に用量依存的な影響を及ぼさなかった。抗HIV療法を受けている1793例のHIV感染患者では,アタザナビル及び比較対照薬のQTc延長作用は同等であった。アタザナビルを投与された健康成人又はHIV感染患者のいずれにおいても,500msecを超えるQTc間隔は認められなかった。
アタザナビル400mg1日1回とCYP3Aの基質であるジルチアゼム180mg1日1回投与の臨床薬理試験において,PR間隔に対して相加的な影響が認められた。また,アタザナビル400mg1日1回とアテノロール50mg1日1回投与の臨床薬理試験において,PR間隔に対して相加的な影響は認められなかった。(外国人における成績)[9.1.1,10.2参照]
注)承認最大用量は400mgである。
18. 薬効薬理
18.1 作用機序
アタザナビル(ATV)はアザペプチド系のHIV-1プロテアーゼ阻害薬(PI)である。
HIV-1に感染した細胞において,本剤はプロテアーゼ阻害作用によりHIVウイルスの構造蛋白(Gag-Pol)に影響を及ぼし,その結果,感染性を有する成熟ウイルスの産生を抑制する。
18.2 抗ウイルス作用(in vitro試験)
末梢血単核細胞,マクロファージ,CEM-SS細胞及びMT-2細胞に感染させた各種HIV-1分離株におけるATVの抗ウイルス作用のEC50値は,ヒト血清非存在下で2〜5nMであった。ATVは細胞培養試験において,HIV-1グループM サブタイプ A,B,C,D,AE,AG,F,G及びJの分離株に対して活性を示した。HIV-2分離株に対してはEC50値1.9〜32nMの変動のある活性を示した。細胞培養試験において,ATVと非核酸系逆転写酵素阻害薬(NNRTI:デラビルジン,エファビレンツ及びネビラピン),PI(アンプレナビル,インジナビル,ロピナビル,ネルフィナビル,リトナビル及びサキナビル),逆転写酵素阻害薬(NRTI:アバカビル,ジダノシン,エムトリシタビン,ラミブジン,サニルブジン,テノホビル,ザルシタビン及びジドブジン),ウイルス感染治療薬であるアデホビル及びリバビリンのいずれかとの2剤併用の抗ウイルス活性試験では,併用薬剤間で拮抗作用は認められず,細胞毒性の増強もなかった。
18.3 薬剤耐性
18.3.1 細胞培養試験
細胞培養試験において,ATVに5ヵ月間曝露した場合,ATVに対する感受性が1/183〜1/93に低下した3種の異なるウイルス株が得られた。これらのATV耐性にはHIV-1ウイルスのI50L,N88S,I84V,A71V及びM46Iのアミノ酸置換が関与していた。また,アミノ酸置換はプロテアーゼ開裂部位でも認められた。I50Lを有し,PI関連の他のメジャーなアミノ酸置換を含まない組み換えウイルスでは,細胞培養試験において増殖障害が認められ,また他のPI(アンプレナビル,インジナビル,ロピナビル,ネルフィナビル,リトナビル及びサキナビル)に対する感受性増大が認められた。ATV及びアンプレナビルの選択的耐性置換としてI50LとI50Vがそれぞれ認められたが,これらに交叉耐性はなかった。
18.3.2 治療経験のない患者での臨床試験
(1)
治療経験のないHIV感染患者にATV300mg1日1回+RTV100mg1日1回(ATV/RTV群)及びATV400mg1日1回(ATV群)のラミブジン+徐放性サニルブジン併用下における比較試験(AI424-089試験)
| ATV/RTV群 |
ATV群 |
|
|---|---|---|
| 96週時ウイルス学的失敗数 (≥50copies/mL) |
15 (16%) |
34 (32%) |
| 遺伝子型及び表現型変化を有する ウイルス学的失敗数 |
5 | 17 |
| 96週時ATV耐性の ウイルス学的失敗分離株 |
0/5 (0%)b |
4/17 (24%)b |
| 96週時I50L変異を有する ウイルス学的失敗分離株c |
0/5 (0%)b |
2/17 (12%)b |
| 96週時ラミブジン耐性を有する ウイルス学的失敗分離株 |
2/5 (40%)b |
11/17 (65%)b |
a:ウイルス学的失敗は96週間ウイルス量が減少しなかった患者及び96週時にウイルス学的リバウンドを生じた患者あるいはウイルス量減少不十分により治療を中止した患者を含む。
b:遺伝子型及び表現型データが存在するウイルス学的失敗分離株の割合(%)。
c:I50I/Lの混合変化が他のATV400mg投与患者2例で認められたが,いずれもATVに対する表現型耐性は認められなかった。
(2)治療経験のないHIV感染患者にATVの300mg1日1回+RTV100mgを投与した試験(AI424-138試験)
ATV/RTVの96週間治療中にウイルス学的失敗(≥400copies/mL)を経験した患者あるいはウイルス量減少が基準値到達前に投与を中止した患者の血液サンプルについて遺伝子型及び表現型解析を実施した。解析数は39例(9%)であった。その結果,ATV/RTV治療グループにおいて,ウイルス学的失敗分離株の1例ではATVに対する感受性が1/56に低下し,L10F,V32I,K43T,M46I,A71I,G73S,I85I/V及びL90MのPI関連の置換が認められた。また,治療失敗分離株5例では,M184I(1例)又はM184V(4例)の置換を有するエムトリシタビン耐性が発現した。
(3)治療経験のないHIV感染患者にATVの400mgを1日1回投与した試験
ATV400mgのみの治療でウイルス学的失敗を経験し,耐性となった患者からの分離株では多くの場合I50Lの置換が認められ(ATV平均治療期間50週間),またA71Vの置換を合併していた。また,1ヵ所以上のPI関連の置換(例えばV32I,L33F,G73S,V82A,I85V又はN88S)がI50Lと同時にあるいはI50Lなしに発現していた。
未治療患者において,主要なPI関連置換を有さずにI50Lの置換のみが発現したウイルス分離株は,ATVに対する表現型耐性を示したが,他のPI(アンプレナビル,インジナビル,ロピナビル,ネルフィナビル,リトナビル及びサキナビル)に対しては細胞培養試験で感受性の保持が観察された。
18.3.3 治療経験を有する患者での臨床試験
治療経験を有するHIV-1患者にATV又はATV/RTVを投与した試験。ウイルス学的失敗を経験した患者から分離したほとんどのATV耐性分離株では,複数のPIに対する耐性と関連したアミノ酸置換が発現し,PIに対する感受性低下が認められた。ATV300mgとRTV100mgの1日1回(同時にテノホビルと1種のNRTI)治療で失敗した患者のウイルス分離株において,最も一般的に認められた置換は,V32I,L33F/V/I,E35D/G,M46I/L,I50L,F53L/V,I54V,A71V/T/I,G73S/T/C,V82A/T/L,I85V及びL89V/Q/M/Tであった。その他の置換として,E34K/A/Q,G48V,I84V,N88S/D/T,及びL90Mが10%未満の患者分離株で認められた。概してATV又はATV/RTV投与開始前の患者のHIV-1ウイルスにI50Lの置換を含む複数のPI耐性置換が存在する場合には,ATVにも耐性が生じた。I50L置換は,治療経験を有する患者にATVの長期投与後にウイルス学的失敗を経験した患者でも確認されている。ATV治療によりプロテアーゼ開裂部位の変化も生じたが,これらの出現はATV耐性の程度とは相関しなかった。
18.4 交差耐性
PIの治療経験を有する患者にATVを投与した臨床試験において,ATV投与前にウイルス分離株の表現型及び遺伝子型解析を実施した結果,複数のPIに交差耐性を示し,ATVに対しても交差耐性を示した。I84V又はG48V置換を有する分離株の90%以上が,ATVに耐性を示した。また,L90M,G73S/T/C,A71V/T,I54V,M46I/LあるいはV82に置換を有する分離株の60%以上がATV耐性であり,更に他の置換に加えてD30Nを有する分離株の38%がATV耐性であった。ATVに耐性を示す分離株は他のPIに対しても交差耐性を示し,インジナビル,ロピナビル,ネルフィナビル,リトナビル及びサキナビルに対しては90%以上の分離株が,また,アンプレナビルに対しては80%が耐性を示した。治療経験を有する患者において,PI耐性に関連するアミノ酸置換に加えてI50Lを発現したPI耐性ウイルス分離株は,他の複数のPIに対しても交差耐性を示した。
19. 有効成分に関する理化学的知見
| 一般名 | アタザナビル硫酸塩(Atazanavir Sulfate) |
|---|---|
| 化学名 | Dimethyl(3S,8S,9S,12S)-9-benzyl-3,12-di-tert-butyl-8-hydroxy-4,11-dioxo-6-[4-(pyridin-2-yl)benzyl]-2,5,6,10,13-pentaazatetradecanedioate monosulfate |
| 分子式 | C38H52N6O7・H2SO4 |
| 分子量 | 802.93 |
| 構造式 | 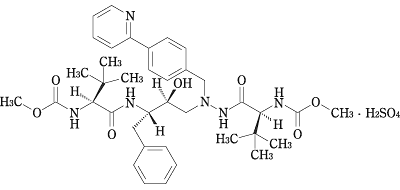 |
| 性状 | アタザナビル硫酸塩は白色〜微黄色の粉末である。メタノール又はエタノール(95)に溶けやすく,水に溶けにくい。 |
| 分配係数 |
646(1-オクタノール/水) |
20. 取扱い上の注意
開封後は湿気を避けて保存すること。
22. 包装
〈レイアタッツカプセル150mg〉
60カプセル[瓶,バラ]
〈レイアタッツカプセル200mg〉
60カプセル[瓶,バラ]
23. 主要文献
- JEAN-MICHEL MOLINA et al.: J Acquir Immune Defic Syndr. 2010; 53(3): 323-332
- KATHLEEN SQUIRES et al.: J Acquir Immune Defic Syndr. 2004; 36(5):1011-1019
- MARGARET JOHNSON et al: AIDS. 2006; 20(5): 711-718
24. 文献請求先及び問い合わせ先
ブリストル・マイヤーズ スクイブ株式会社 メディカル情報グループ
(住所)東京都新宿区西新宿6-5-1
(TEL)0120-093-507
26. 製造販売業者等
26.1 製造販売元
ブリストル・マイヤーズ スクイブ株式会社
東京都新宿区西新宿6-5-1