ストックリン錠200mg/600mgの添付文書
- 商品名:
- ストックリン錠200mg/600mg
- 一般名:
- エファビレンツ
- 略称 :
- EFV

添付文書の読み方
ここで提供している添付文書情報は、2021年12月現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
抗ウイルス化学療法剤
エファビレンツ
- *2021年7月改訂(第2版)
2020年2月改訂(第1版) - 劇薬
処方箋医薬品注):注意-医師等の処方箋により使用すること
| 日本標準商品分類番号 | 87625 |
|---|
| 貯法 | 室温保存 |
|---|---|
| 有効期間 | 3年 |
| 200mg | 600mg | |
|---|---|---|
| 承認番号 | 22100AMX00490000 | 22000AMX01554000 |
| 販売開始 | 2009年11月 | 2008年6月 |
2. 禁忌(次の患者には投与しないこと)
2.1
本剤の成分に対し過敏症の既往歴のある患者
2.2
トリアゾラム、ミダゾラム、エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン、ジヒドロエルゴタミンメシル酸塩、メチルエルゴメトリンマレイン酸塩及びエルゴメトリンマレイン酸塩を投与中の患者[10.1参照]
2.3
ボリコナゾールを投与中の患者[10.1参照]
2.4
シメプレビル、アスナプレビル、ダグラタスビル塩酸塩・アスナプレビル・ベクラブビル塩酸塩を投与中の患者[10.1参照]
2.5
エルバスビル、グラゾプレビルを投与中の患者[10.1参照]
3. 組成・性状
3.1 組成
| 販売名 | ストックリン®錠200mg | ストックリン®錠600mg |
|---|---|---|
| 有効成分 | エファビレンツ | |
| 分量 | 200mg | 600mg |
| 添加剤 | クロスカルメロースナトリウム、結晶セルロース、ラウリル硫酸ナトリウム、ヒドロキシプロピルセルロース、乳糖水和物、ステアリン酸マグネシウム、ヒプロメロース、酸化チタン、マクロゴール400、黄色三二酸化鉄、カルナウバロウ | |
3.2 製剤の性状
| 販売名 | ストックリン®錠200mg | ストックリン®錠600mg | |
|---|---|---|---|
| 剤形・色調 | 円形、フィルムコーティング錠、黄色 | 長円形、フィルムコーティング錠、黄色 | |
| 外形 | 表面 |  直径:11.1mm |
 長径:19mm、 短径:9.5mm |
| 裏面 |  |
 |
|
| 側面 |  厚さ:4.3mm |
 厚さ:7.5mm |
|
| 識別コード | 223 | 225 | |
4. 効能又は効果
HIV-1感染症
6. 用法及び用量
通常、成人にはエファビレンツとして600mgを1日1回経口投与する。本剤は、食事の有無にかかわらず投与できる。なお、投与に際しては必ず他の抗HIV薬と併用すること。
7. 用法及び用量に関連する注意
7.1
本剤は、単独で投与しないこと。また、他の治療が無効の場合に本剤を単独で追加投与しないこと。本剤による治療は、患者に未投与の1種類以上の抗レトロウイルス薬(ヌクレオシド系逆転写酵素阻害剤又はHIVプロテアーゼ阻害剤)との併用により開始すること。本剤と併用する抗レトロウイルス薬の選択にはウイルスの交差耐性の可能性を考慮すること。本剤を単独療法として投与する場合、耐性ウイルスが急速に出現する。
7.2
薬剤への忍容性がないために併用療法中の抗レトロウイルス薬の投与を中断する場合は、すべての抗レトロウイルス薬を同時に中止するよう十分に考慮すること。不忍容の症状が消失した際はすべての抗レトロウイルス薬の投与を同時に再開すること。抗レトロウイルス薬の間欠的単独療法及び逐次的再導入は、薬剤耐性突然変異ウイルスの出現の可能性が増加するため望ましくない。
7.3
神経系の副作用の忍容性を改善するため、治療当初の2~4週間及び神経系の副作用が継続する患者では、就寝時の投与が推奨される。[8.3参照]
7.4
食物との併用により、本剤の曝露量を増加させ、副作用の発現頻度を増加させるおそれがある。本剤は、食事の有無にかかわらず投与できるが、空腹時、可能な限り就寝時の服用が望ましい。[16.2.1参照]
7.5
何らかの理由により本剤の投与を中断する場合は、他の抗レトロウイルス薬の投与中止を十分に考慮すること。同様に、併用している抗ウイルス療法が一時的に中止される場合は、本剤の投与も中止すること。すべての抗レトロウイルス薬の投与を同時に再開すること。
7.6
リファンピシンと併用投与する場合、本剤の投与量を800mg/日に増加すること。[10.2参照]
8. 重要な基本的注意
*8.1
本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又はそれに代わる適切な者に、次の事項についてよく説明し、同意を得た後、使用すること。
- 本剤はHIV感染症の根治療法薬ではないことから、日和見感染を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。
- 本剤の長期投与による影響については、現在のところ不明である。
- 抗HIV療法による効果的なウイルス抑制は、性的接触による他者へのHIV感染の危険性を低下させることが示されているが、その危険性を完全に排除することはできないこと。
- 抗HIV療法が、血液等による他者へのHIV感染の危険性を低下させるかどうかは証明されていないこと。
- 本剤は、処方通りに毎日服用すること。本剤は、常に他の抗レトロウイルス薬と併用しなければならないこと。また、担当医への相談なしで、用量を変更したり治療を中止しないこと。
- 本剤は他の薬剤と相互作用を示す可能性があるので、他の薬剤の服用の有無について担当医に報告すること。
- 本剤をアルコール又は中枢神経作用薬と併用するとき、相加的に中枢神経系効果が増強されるので注意すること。
- 本剤はめまい、集中力障害、嗜眠状態を引き起こすことがある。これらの症状があらわれた場合は、自動車の運転や機械の操作等、潜在的な危険のある労働を避けること。
- 動物実験で胎児に奇形が認められているため、本剤を投与中及び投与中止後12週間を経過していない女性は、適切な避妊方法を用いて妊娠を避けるようにし、妊娠した場合は担当医に報告すること。
8.2
本剤に関する臨床試験において軽・中等度の発疹が報告されており、一般に投与開始2週間以内に発現し、通常は投与継続中に1ヵ月以内で消失する。適切な抗ヒスタミン薬もしくはコルチコステロイドの投与が忍容性の改善を促し、発疹の消失を早めることがある。[11.1.1参照]
8.3
対照群患者及び本剤投与群患者両群ともに重度の急性うつ病(自殺願望及び自殺企図を含む)がまれに報告された。妄想、不穏当な行動及び重度の急性うつ病(自殺願望及び自殺企図を含む)が発現した患者には、本剤の投与中止を考慮すること。本剤を投与している患者の52%に精神神経系症状が報告された。これらの症状の主なものは、めまい、集中力障害、傾眠、異夢及び不眠であった。比較対照臨床試験では、これらの症状は本剤1日600mgを投与された患者の2.6%、対照群の患者の1.4%で重度であった。臨床試験では、本剤を投与された患者の2.6%が精神神経系症状のために投与を中止した。精神神経系症状は一般に投与開始1~2日後に発現し、通常は投与継続中に2~4週間で消失する。[7.3、9.1.2、11.2参照]
8.4
重篤な肝障害が報告されているため、本剤を投与する際には、肝酵素のモニタリングが推奨される。血清トランスアミナーゼの正常範囲の上限より5倍以上の上昇が持続している患者では、本剤による重篤な肝毒性発症のリスクより本剤の有用性が上回ると判断された場合にのみ投与すること。[9.1.1、10.2、11.1.2参照]
8.5
本剤を投与している患者では、脂質のモニタリングを考慮すること。本剤を投与された数名の非感染ボランティアに10~20%の総コレステロール上昇が認められた。また、本剤を非空腹時の患者に投与した際、血清トリグリセライド及びコレステロールの軽微な上昇が認められた。また別の試験では、[本剤+ジドブジン(ZDV)+ラミブジン(3TC)]を48週間投与した患者において、総コレステロール、HDLコレステロール、空腹時LDLコレステロール及び空腹時トリグリセライドのそれぞれベースラインから21%、24%、18%及び23%の上昇が認められた。しかしながら、これらの知見の臨床上の意義は不明である。8.6 本剤を含む抗HIV薬の多剤併用療法を行った患者で、免疫再構築症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
8.7
QT延長があらわれることがあるので、定期的に検査を実施するなど観察を十分に行うこと。[11.1.3、17.3.1参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 B型、C型肝炎感染の既往のある患者あるいはその疑いのある患者
重篤な肝障害発現のリスクが増加する。また、肝機能障害が増悪するおそれがある。[8.4、10.2、11.1.2参照]
9.1.2 精神病あるいは薬物乱用の既往歴のある患者
妄想及び不穏当な行動が報告された(本剤を投与された患者1,000例につき約1~2例)。[8.3参照]
9.3 肝機能障害患者
慢性肝疾患患者に対する臨床試験は実施していない。
9.5 妊婦
妊婦又は妊娠している可能性のある女性には投与しないことが望ましい。
海外の抗HIV薬治療下妊娠症例登録制度において、本剤を妊娠中に服用した妊婦から生まれた新生児に脊髄髄膜瘤等の神経管欠損が報告されている。また、動物実験(カニクイザル)において、胎児/新生児20匹のうち3匹で奇形が認められた。妊娠したサルにエファビレンツ60mg/kg/day(ヒトに600mg/日を投与したときと同様の血漿中薬物濃度を示す用量)を投与した。1胎児において無脳及び片眼の無眼球症が認められた。別の胎児では小眼球症が認められ、第3の胎児では口蓋裂が認められた。
9.6 授乳婦
授乳を避けさせること。
ラットにおける実験では、本剤が乳汁中に排泄されることが認められている。また、ヒトの乳汁中に移行することも認められている1)。
9.8 高齢者
一般に生理機能が低下している。
10. 相互作用
本剤は、チトクロームP450(CYP)3A4及びCYP2B6の誘導剤である。CYP3A4もしくはCYP2B6の基質である他の化合物の血中濃度は、本剤との併用により低下することがある。[16.4.3参照]
*10.1 併用禁忌(併用しないこと)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| トリアゾラム: ハルシオン *ミダゾラム: ドルミカム ブコラム ミダフレッサ エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン: クリアミン配合錠 ジヒドロエルゴタミンメシル酸塩 メチルエルゴメトリンマレイン酸塩: パルタンM エルゴメトリンマレイン酸塩: エルゴメトリン [2.2参照] |
これらの薬剤の代謝が抑制され、重篤な又は生命に危険を及ぼすような事象(不整脈、持続的な鎮静、呼吸抑制)が起こる可能性がある。 | CYP3A4に対する競合による。 |
| ボリコナゾール: ブイフェンド [2.3参照] |
ボリコナゾールとの併用により、ボリコナゾールのAUC及びCmaxがそれぞれ77%及び61%減少し、本剤のAUC及びCmaxがそれぞれ44%及び38%増加した。 | 機序不明 |
| シメプレビル: ソブリアード アスナプレビル: スンベプラ ダグラタスビル塩酸塩・ アスナプレビル・ ベクラブビル塩酸塩: ジメンシー配合錠 [2.4参照] |
本剤との併用により、これらの薬剤の血漿中濃度が低下し、効果が減弱するおそれがある。 | 本剤のCYP3A4誘導作用により、これらの薬剤の代謝が促進されるおそれがある。 |
| エルバスビル: エレルサ グラゾプレビル: グラジナ [2.5参照] |
本剤との併用により、これらの薬剤の血漿中濃度が低下し、効果が減弱するおそれがある。 | 本剤のCYP3A4及びP-gp誘導作用によりこれらの薬剤の代謝及び排出が促進されるおそれがある。 |
*10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
| インジナビル | インジナビル(800mg、8時間ごと)と本剤を併用して投与した場合、酵素誘導の結果としてインジナビルのAUC及びCmaxがそれぞれ約31%及び16%減少した。 | 本剤のCYP3A4誘導作用により、インジナビルの代謝が促進されるおそれがある。 |
| リトナビル | 非感染ボランティアにおいて本剤600mg(1日1回、就寝時投与)とリトナビル500mg(12時間ごと投与)について試験を行ったとき、併用の忍容性は良好ではなく、高頻度の臨床的有害事象(例:めまい、嘔気、異常感覚)及び臨床検査値異常(肝酵素上昇)が認められた。本剤をリトナビルと併用する場合は肝酵素のモニタリングが推奨される。 | 機序不明 |
| サキナビル | サキナビル(1,200mg1日3回、ソフトゲル製剤)と本剤を併用した場合、サキナビルのAUC及びCmaxがそれぞれ62%及び45~50%減少したとの報告がある。併用するプロテアーゼ阻害剤がサキナビルのみの場合は本剤の使用は推奨されない。 | 機序不明 |
| ホスアンプレナビル | ホスアンプレナビル1,400mg及びリトナビル200mgの1日1回投与と本剤600mg1日1回を併用した場合、アンプレナビルのAUCが13%、Cminが36%低下したが、リトナビルを300mgに増量すると、アンプレナビルの血中濃度は維持された。また、ホスアンプレナビル700mg及びリトナビル100mgの1日2回投与と本剤600mg1日1回を併用した場合、アンプレナビルの血中濃度に著しい変化はなかった。 | 本剤のCYP3A4誘導作用により、アンプレナビルの代謝が促進される。 |
| アタザナビル | 本剤600mgとアタザナビルとの併用により、アタザナビルの曝露量が減少した。本剤をアタザナビルと併用する際には、さらに低用量のリトナビルを併用するとともに、アタザナビルの用量調節が必要である。HIV治療歴のない患者に本剤を併用投与する場合、アタザナビル300mg、リトナビル100mg、本剤600mgを1日1回投与することが推奨される。 HIV治療歴のある患者におけるアタザナビル及び本剤の推奨用量は確立していない。 |
機序不明 |
| ロピナビル・リトナビル | ロピナビル・リトナビル(カプセル剤)と本剤を併用した場合、ロピナビル・リトナビルの単独投与時と比べてロピナビルのCminが39%低下した。 | 本剤のCYP3A4誘導作用により、ロピナビルの代謝が促進されるおそれがある。 |
| ダルナビル | ダルナビル/リトナビル(300mg/100mg1日2回)と本剤(600mg1日1回)を併用した場合、単独投与時と比べてダルナビルのAUC、Cmax及びCminがそれぞれ13%、15%及び31%減少し、本剤のAUC、Cmax及びCminがそれぞれ21%、15%及び17%増加した。またダルナビル/リトナビル(900mg/100mg1日1回)と本剤(600mg1日1回)を併用した場合、ダルナビルのAUC及びCminがそれぞれ14%及び57%減少し、ダルナビルのCmax及び本剤の曝露は変化がなかった。 | 本剤のCYP3A4誘導作用により、ダルナビルの代謝が促進されるおそれがある。 |
| マラビロク | 本剤(600mg経口1日1回)とマラビロク(100mg経口1日2回)を併用した場合、マラビロク単剤投与と比較して、マラビロクのAUC及びCmaxはそれぞれ45%及び51%減少した。 | 本剤のCYP3A4誘導作用によりマラビロクの代謝が促進されるおそれがある。 |
| ドルテグラビル | 本剤(600mg経口1日1回)とドルテグラビル(50mg経口1日1回)を併用した場合、ドルテグラビル単剤投与と比較して、ドルテグラビルのAUC、Cmax及びCminはそれぞれ57%、39%及び75%減少した。 | 本剤のCYP3A4及びUGT1A1誘導作用によりドルテグラビルの代謝が促進されるおそれがある。 |
| *ソホスブビル・ベルパタスビル | 本剤との併用により、ベルパタスビルの血漿中濃度が低下し、ソホスブビル・ベルパタスビルの効果が減弱するおそれがある。 | 本剤のP-gp及びCYP誘導作用により、ベルパタスビルのクリアランスが亢進するおそれがある。 |
| リファンピシン リファブチン [7.6参照] |
非感染ボランティア12例ではリファンピシンにより本剤のAUCが26%、Cmaxが20%減少した。本剤とリファンピシンを併用投与する場合、リファンピシンの用量調節は推奨されない。非感染のボランティアに対する臨床試験において、本剤はリファブチンのCmax及びAUCをそれぞれ32%及び38%低下させた。 | 機序不明 |
| クラリスロマイシン | 本剤(400mg1日1回)とクラリスロマイシン(500mg12時間ごと)を1週間併用した場合、本剤がクラリスロマイシンの薬物動態に対して有意な影響を及ぼした。本剤と併用した場合に、クラリスロマイシンのAUC及びCmaxがそれぞれ39%及び26%減少する一方で、クラリスロマイシン水酸化代謝物のAUC及びCmaxがそれぞれ34%及び49%増加した。このようなクラリスロマイシンの血漿中濃度の変化の臨床上の意義は不明である。非感染ボランティアの46%で本剤とクラリスロマイシンを投与中に発疹が発現した。本剤はクラリスロマイシンと併用投与した場合には用量調節は推奨されない。クラリスロマイシンの代替薬を考慮すること。 | 機序不明 |
| 経口避妊薬: レボノルゲストレル・エチニルエストラジオール レボノルゲストレル |
本剤(600mg1日1回)と経口避妊薬(エチニルエストラジオール0.035mg・ノルゲスチメート0.25mg1日1回)を14日間併用した場合、本剤はエチニルエストラジオールの血漿中濃度に影響を与えなかったが、ノルゲスチメートの活性代謝物であるノルエルゲストロミン及びレボノルゲストレルのAUCはそれぞれ64%及び83%減少した。これらの作用の臨床上の意義は不明である。一方、本剤の血漿中濃度への影響は認められなかった。本剤と経口避妊薬の併用による相互作用の可能性は十分に検討されていない。経口避妊薬に加えて信頼できる防御的避妊法(コンドーム)を用いること。 | 機序不明 |
| セイヨウオトギリソウ(St. John's Wort、セント・ジョーンズ・ワート)含有食品 | 本剤の血中濃度が低下し、抗ウイルス作用の欠如及び本剤又は他の非ヌクレオシド系逆転写酵素阻害剤の耐性化が起こるおそれがあるので、本剤投与時はセイヨウオトギリソウ含有食品を摂取しないよう注意すること。 | セイヨウオトギリソウにより誘導された肝薬物代謝酵素(CYP3A4)が本剤の代謝を促進し、クリアランスを上昇させるためと考えられている。 |
| アトルバスタチン | 非感染ボランティアにおいて、本剤(600mg経口1日1回)とアトルバスタチン(10mg経口1日1回)を併用した場合、アトルバスタチン単剤投与と比較して、定常状態におけるアトルバスタチン及びその由来物質のAUC及びCmaxを減少させた(アトルバスタチン:43%及び12%、2-ヒドロキシアトルバスタチン:35%及び13%、4-ヒドロキシアトルバスタチン:4%及び47%、総HMG-CoA還元酵素阻害活性物質:34%及び20%)。 | 機序不明 |
| プラバスタチン | 非感染ボランティアにおいて、本剤(600mg経口1日1回)とプラバスタチン(40mg経口1日1回)を併用した場合、プラバスタチン単剤投与と比較して、定常状態におけるプラバスタチンのAUC及びCmaxが40%及び18%減少した。 | 機序不明 |
| シンバスタチン | 非感染ボランティアにおいて、本剤(600mg経口1日1回)とシンバスタチン(40mg経口1日1回)を併用した場合、シンバスタチン単剤投与と比較して、定常状態におけるシンバスタチン及びその由来物質のAUC及びCmaxを減少させた(シンバスタチン:69%及び76%、シンバスタチンのオープンアシド体:58%及び51%、HMGCoA還元酵素阻害活性物質:60%及び62%、総HMG-CoA還元酵素阻害物質:60%及び70%)。 | 本剤のCYP3A4誘導作用により、シンバスタチンの代謝が促進されるおそれがある。 |
| 抗痙攣薬: カルバマゼピン |
非感染ボランティアにおいて、本剤(600mg経口1日1回)とカルバマゼピン(400mg1日1回)を併用した場合、定常状態におけるカルバマゼピンのAUC、Cmax、Cminはそれぞれ27%、20%、35%減少し、本剤のAUC、Cmax、Cminはそれぞれ36%、21%、47%減少した。定常状態における活性型カルバマゼピンエポキシド代謝物のAUC、Cmax、Cminは変化がなかった。カルバマゼピンの血漿中濃度は定期的にモニタリングすべきである。 フェニトイン、フェノバルビタール、あるいはチトクロームP450で代謝される他の抗痙攣薬との相互作用についてのデータは得られていない。本剤がこれらの薬剤と併用して投与される場合、各薬剤の血漿中濃度を増加あるいは減少させる可能性があるので、血漿中濃度を定期的にモニタリングすべきである。 |
機序不明 |
| イトラコナゾール | 非感染ボランティアにおいて、本剤(600mg経口1日1回)とイトラコナゾール(200mg経口12時間ごと)を併用した場合、イトラコナゾール単剤投与と比較して、定常状態におけるイトラコナゾールのAUC、Cmax及びCminはそれぞれ39%、37%及び44%減少し、ヒドロキシイトラコナゾールのAUC、Cmax及びCminはそれぞれ37%、35%及び43%減少した。 | 機序不明 |
| *ポサコナゾール | 本剤(400mg経口1日1回)とポサコナゾール(400mg経口1日2回)を併用した場合、単独投与時と比べてポサコナゾールのAUC及びCmaxがそれぞれ50%及び45%低下した。治療上の有益性が危険性を上回る場合を除き、併用は避けること。やむを得ず併用する場合は、真菌症の発症の有無を注意深くモニタリングするなど患者の状態を慎重に観察すること。 | 本剤との併用により、ポサコナゾールのクリアランスが亢進し、ポサコナゾールの血漿中濃度が低下する。 ポサコナゾールが基質となるUGT1A4及び/又はP-gpに対する本剤の誘導作用が関与している可能性がある。 |
| ジルチアゼム | 非感染ボランティアにおいて、本剤(600mg経口1日1回)とジルチアゼム(240mg経口1日1回)を併用した場合、ジルチアゼム単剤投与と比較して、定常状態におけるジルチアゼムのAUC、Cmax及びCminはそれぞれ69%、60%及び63%減少し、デスアセチルジルチアゼムのAUC、Cmax及びCminは75%、64%及び62%減少し、NモノデスメチルジルチアゼムのAUC、Cmax及びCminは37%、28%及び37%減少した。 | 機序不明 |
| テラプレビル | テラプレビル(750mg、8時間ごと)と本剤(600mg1日1回)を併用した場合、テラプレビルのAUC、Cmax及びCminが単剤投与時と比べてそれぞれ26%、9%及び47%減少した。 | 本剤のCYP3A4誘導作用により、テラプレビルの代謝が促進されるおそれがある。 |
| アトバコン・プログアニル | 本剤(600mg1日1回)とアトバコン・プログアニル(250mg/100mg単回投与)を併用した場合、アトバコンのAUC及びCmaxはそれぞれ75%及び44%、プログアニルのAUCは43%低下した。 | 機序不明 |
| 肝毒性が知られている薬剤: アセトアミノフェンフェニトイン リファンピシン等 [8.4、9.1.1、11.1.2参照] |
重篤な肝障害発現のリスクが増加する。 | 機序不明 |
11. 副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1 重大な副作用
11.1.1 皮膚粘膜眼症候群(Stevens-Johnson症候群)(0.1%未満)、多形紅斑(0.1%未満)
本剤投与患者の1%未満で、水疱、湿性の落屑又は潰瘍形成を随伴した重度の発疹が報告されている。水疱、落屑、粘膜波及又は発熱に関連する重度の発疹が発現した患者では本剤の投与を中止すること。[8.2参照]
11.1.2 肝不全(頻度不明)
重篤な肝障害があらわれることがある。[8.4、9.1.1、10.2参照]
11.1.3 QT延長(頻度不明)
[8.7、17.3.1参照]
11.2 その他の副作用
| 10%以上 | 1~10%未満 | 1%未満 | 頻度不明 | |
|---|---|---|---|---|
| 全身性一般 | 頭痛、インフルエンザ様症候群、疼痛 | 無力症、倦怠感、発熱 | アルコール不耐性、ほてり、失神、末梢性浮腫 | 体脂肪の再分布/蓄積(後頸部、胸部、腹部、後腹膜等の部位) |
| 消化器 | 嘔気、嘔吐、下痢、消化不良 | 胃炎、胃腸炎、胃食道逆流、アミラーゼ上昇、口渇、腹痛、鼓腸放屁、食欲亢進、食欲不振 | 膵炎 | |
| 心・血管系 | 潮紅、動悸、頻脈 | |||
| 肝臓 | AST上昇、ALT上昇、γ-GTP上昇 | 肝炎、総ビリルビン上昇 | ||
| 筋・骨格系 | 関節痛、筋痛 | |||
| 精神神経系注1) | めまい、不眠、集中力障害、疲労 | うつ症状悪化、激越、健忘、不安、運動失調、感情不安定、多幸症、幻覚、偏頭痛、異常感覚、抑うつ、神経過敏、傾眠、異夢、錯乱 | 協調障害、インポテンス、性欲減退、性欲亢進、神経痛、末梢神経障害、言語障害、痙攣、離人症、思考異常、振戦 | 感情鈍麻、精神病、小脳障害(平衡障害、眼振等)、カタトニー |
| 呼吸器 | 喘息、副鼻腔炎、上気道感染 | |||
| 皮膚 | 発疹、斑状丘疹性皮疹、紅斑 | 脱毛、湿疹、脂漏、じん麻疹、毛包炎、瘙痒、多汗、多汗(夜間) | 痤瘡 | 皮膚剝離、光線過敏性皮膚炎 |
| その他 | 好中球減少、耳鳴、血糖値上昇、体重減少、視力異常、味覚倒錯 | 総コレステロール上昇、血清トリグリセライド上昇、体重増加、複視、嗅覚錯誤 | 女性化乳房、貧血(赤血球数減少、ヘモグロビン低下等) |
注1)[8.3参照]
12. 臨床検査結果に及ぼす影響
本剤は、カンナビノイドレセプターに結合しないが、本剤投与時に複数の尿カンナビノイド試験で、偽陽性が認められている。
13. 過量投与
13.1 処置
本剤は蛋白質との結合率が高いため、透析では血液から薬物を有意に除去する可能性は低い。[16.3参照]
16. 薬物動態
16.1 血中濃度
16.1.1
健康成人男子に200~600mg(カプセル)を空腹時に単回経口投与した場合、本剤の血漿中濃度は、投与後2.5~3.5時間にピークに達し、半減期は約56.4~58.3時間であった(各n=6)。血漿中濃度曲線下面積(AUC)及び最高血漿中濃度(Cmax)は用量依存的に上昇し、600mg単回経口投与におけるAUCは316.9±69.8μM・h、Cmaxは6.8±2.3μMであった(n=6)。(図1)
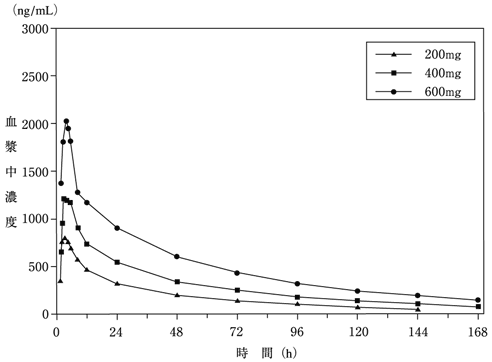
(換算値:1,000ng/mL=3.17μM)
16.1.2
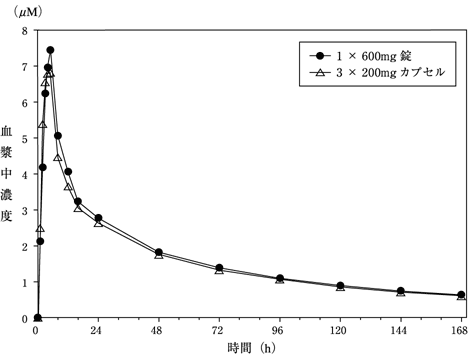
健康成人(n=21)に600mg錠1錠及び600mg(カプセル)を空腹時に単回経口投与し、両製剤の生物学的同等性について検討した。カプセル投与に対する600mg錠投与の幾何平均比及び90%信頼区間は、AUC0-tで1.02(0.96-1.09)、Cmaxで1.10(0.99-1.23)であり、両製剤は生物学的に同等であることが確認された(外国人データ)。(表、図2)
| 用量及び剤形 | AUC0-t (μM・h) |
Cmax (μM) |
Tmaxa) (h) |
t1/2b) (h) |
|---|---|---|---|---|
| 600mg錠剤 | 338.77±111.37 | 8.06±1.95 | 4(2-8) | 78.21±27.74 |
| 200mgカプセル×3 | 326.97±112.47 | 7.50±2.81 | 4(2-5) | 75.81±29.56 |
平均±標準偏差(n=21)
a)中央値(範囲)
b)調和平均±ジャックナイフ標準偏差
16.1.3
非感染ボランティアに100~1,600mg(カプセル)を単回経口投与後、本剤の血漿中濃度は投与5時間以内に1.6~9.1μMのCmaxに達した。Cmax及びAUCの用量依存的な増加は、1,600mgまでの用量で認められたが、上昇は完全には用量に比例せず、高用量での吸収の低下が示唆された。反復投与後もTmax(3~5時間)に変化はなく、血漿中濃度は投与6~7日で定常状態に到達した(外国人データ)。
16.1.4
HIV感染患者における定常状態でのCmax、Cmin及びAUCは、1日量200~600mg(カプセル)の範囲で線形性が認められた。HIV感染患者(n=35)に600mg(カプセル)を1日1回反復経口投与した場合、定常状態におけるCmaxは12.9μM、Cminは5.6μM、AUCは184μM・hであった(外国人データ)。
16.2 吸収
16.2.1 食事の影響
非感染ボランティアに600mg錠1錠を高脂肪食(約1,000kcal、カロリーの50~60%が脂肪由来)摂取後単回経口投与した場合、空腹時投与時に比べて本剤のAUCは28%、Cmaxは79%上昇することが認められた。Tmax及び半減期は食事摂取の有無における有意な差は認められなかった(外国人データ)。
非感染ボランティアに600mg(カプセル)を高脂肪食(894kcal、脂肪54g、カロリーの54%が脂肪由来)及び低脂肪食(440kcal、脂肪2g、カロリーの4%が脂肪由来)摂取後単回経口投与した場合には、空腹時投与時に比べて、本剤のAUCはそれぞれ22%及び17%、Cmaxはそれぞれ39%及び51%上昇することが認められた(外国人データ)。[7.4参照]
16.3 分布
本剤はヒト血漿蛋白(主にアルブミン)と強く結合した(約99.5~99.75%)。本剤1日1回200~600mgを1ヵ月間以上投与したHIV-1感染患者9例において、脳脊髄液中濃度は血漿中濃度の0.26~1.19%(平均0.69%)であった。この割合は、血漿中の本剤の非蛋白結合(遊離)画分の約3倍であった(外国人データ)。[13.1参照]
16.4 代謝
16.4.1
ヒトでのin vivo試験及びヒト肝ミクロソームを用いたin vitro試験では、本剤は主にチトクロームP450によって水酸化され、続いてこれら水酸化代謝物はグルクロン酸抱合を受けることが示唆された。これらの代謝物はHIV-1に対して本質的に不活性であった。ヒトにおける本剤の代謝は、CYP3A4及びCYP2B6が関与する。また、臨床用量の血漿中濃度範囲(Ki値;8.5~17μM)で、CYP2C9、CYP2C19及びCYP3A4を阻害するが、CYP2E1は阻害せず、臨床的に到達可能な量よりかなり高濃度でCYP2D6及びCYP1A2(Ki値;82~160μM)を阻害した。
16.4.2
CYP2B6では、516G>T(アミノ酸配列:Gln172His)の一塩基多型が認められている。HIV感染患者に本剤600mg1日1回を含む併用療法を行った場合、CYP2B6の遺伝子型が516TTの患者群では516GGの患者群と比べて、定常状態におけるAUCが約3倍に増加した2)(外国人データ)。なお、CYP2B6 516TTの遺伝子型をもつ集団の割合は、日本人では8.9%との報告がある3)。
16.4.3
本剤はチトクロームP450を誘導し、自己代謝も誘導するため、1日200~400mgを10日間反復投与した結果、AUCは22~42%減少し、t1/2は単回投与時のt1/2(52~76時間)に比べて短縮(40~55時間)した。薬物動態学的相互作用試験で1日400mg及び600mgをインジナビルと併用投与した場合に、200mg投与時と比較してインジナビルのAUCの更なる減少がみられなかったため、CYP3A4誘導の程度は400mgと600mgの用量で同様であると予測される(外国人データ)。[10.参照]
16.5 排泄
16.5.1
健康成人男子に600mg(カプセル)を空腹時に単回経口投与した場合、投与後24時間までの尿中回収率は0.01%以下であった(n=6)。
16.5.2
[14C]エファビレンツを投与したとき、投与した放射能の約14~34%が尿中に回収され、未変化体の排泄は投与量の1%未満であった(外国人データ)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第III相試験(試験006)
ラミブジン(3TC)、HIVプロテアーゼ阻害剤(PI)及び非ヌクレオシド系逆転写酵素阻害剤(NNRTI)による治療歴のないHIV感染患者1,266例を対象に、[本剤(600mg、1日1回)+ジドブジン(ZDV:300mg、12時間ごと)+3TC(150mg、12時間ごと)]、[本剤(600mg、1日1回)+インジナビル(IDV:1,000mg、8時間ごと)]、又は[IDV(800mg、8時間ごと)+ZDV(300mg、12時間ごと)+3TC(150mg、12時間ごと)]を無作為化オープン試験により投与した。ロシュ社製RT-PCR(AmplicorTM)HIV-1アッセイ法を用いたときの血漿中HIV-RNA 400copies/mL未満の患者割合を主要有効性評価項目とした。その結果、本剤を含む3剤併用群でHIV-RNA量が400copies/mL未満に減少した患者の割合は48週で69%、168週で48%であった。168週での成績は他の併用群に比し有意に優れていた。なお、本剤を含む3剤併用群でHIV-RNA量が50copies/mL未満に減少した患者の割合は、48週で64%、168週で42%であった。
17.1.2 海外第III相試験(試験020)
ヌクレオシド系逆転写酵素阻害剤(NRTI)の治療歴があり、PI及びNNRTI治療歴のないHIV感染者327例を対象に[本剤(600mg、1日1回)+IDV(1,000mg、8時間ごと)+2種類のNRTI]又は[IDV(800mg、8時間ごと)+2種類のNRTI]を無作為化二重盲検比較試験により24週間投与した。ロシュ社製RT-PCR(AmplicorTM)HIV-1アッセイ法を用いたときの血漿中HIV-RNA 400copies/mL未満の患者割合を主要有効性評価項目とした。その結果、投与24週後に本剤併用群では68%の患者でHIV-RNA量が400copies/mL未満に減少した。この成績は対照群に比し有意に優れていた。
17.1.3 海外第II相試験(試験ACTG364)
NRTI治療歴のあるHIV感染者195例を対象に[本剤(600mg、1日1回)+ネルフィナビル(NFV:750mg、1日3回)+2NRTI]、[本剤(600mg、1日1回)+2NRTI]又は[NFV+2NRTI]を無作為化二重盲検比較試験により48週間投与した。ロシュ社製RT-PCR(AmplicorTM)HIV-1アッセイ法を用いたときの血漿中HIV-RNA 500copies/mL未満の患者割合を主要有効性評価項目とした。その結果、本剤を含む4剤併用群では70%の患者でHIV-RNA量が500copies/mL未満に減少した。この成績は他の併用群に比し有意に優れていた。
17.3 その他
17.3.1 QT間隔に対する影響
健康成人55例を対象に本剤600mgを1日1回14日間反復経口投与し、QTcF間隔に及ぼす影響を検討した結果、エファビレンツの血漿中濃度とQTcF延長に正の相関関係が認められた。プラセボ補正したQTcF間隔のベースラインからの変化の予測値(90%両側信頼区間の上限)は、CYP2B6の遺伝子型がCYP2B6*1/*1(19例)、*1/*6(19例)、*6/*6(17例)の被験者のCmax幾何平均値でそれぞれ5.6(7.3)ms、6.2(8.0)ms、8.7(11.3)msであった[プラセボ及び陽性対照(モキシフロキサシン400mg1日1回)を用いたクロスオーバー試験](外国人データ)。[8.7、11.1.3参照]
18. 薬効薬理
18.1 作用機序
本剤は、ヒト免疫不全ウイルス1型(HIV-1)の選択的非ヌクレオシド系逆転写酵素阻害剤である。本剤は、HIV-1逆転写酵素(RT)のテンプレート(鋳型)、プライマー又はヌクレオシド三リン酸に対する非拮抗的阻害剤であり、混合型非拮抗阻害形式を示し、拮抗的阻害作用をわずかに併せ持つ。
本剤は、臨床における血中濃度を十分に上回る濃度においても、HIV2RT及びヒトDNAポリメラーゼα、β、γ及びδを阻害しない。
18.2 In vitro抗ウイルス作用
HIV-1のエファビレンツに対するin vitroの感受性の臨床上の意義は確立されていない。末梢血単核細胞(PBMCs)、マクロファージ/単球培養及びPBMCs由来のリンパ芽球細胞株について、エファビレンツのin vitroの抗ウイルス活性の評価を行った。野生型実験室適応菌株及び臨床分離株に対するエファビレンツの90-95%阻害濃度(IC90-95)は、1.7から25nM以下に及んだ。本剤は培養細胞中のHIV-1に対して、NRTIのZDV又はddI、あるいはPIであるIDVとの相乗効果を示した。
18.3 薬剤耐性
S48T、V108I、V179D、Y181C、P236Lの突然変異株、又はプロテアーゼ遺伝子のアミノ酸置換による変異株に対するエファビレンツの効力は、野生型に対して認められたものと同様であった。A98G、K101E、V106A、Y188C又はG190Aの突然変異を含む変異株に対してわずかな耐性(9倍未満)が認められた。in vitroでのエファビレンツ阻害に対する見かけ上の耐性が最も強かった点突然変異は、L100I(17~22倍の耐性)及びK103N(18~33倍の耐性)であった。以下に示すようなRTsをコードする塩基対の1つ以上のアミノ酸置換による変異株、野生型についてはin vitroのエファビレンツに対する耐性の上昇を示した:S48T+G190S(97倍)、Y181C+K103N(133倍)、G190A+K103N(130倍)、Y188L(140~500倍)、K101E+K103N(500倍)、L100I+K103N(>1,000倍)。
IDVあるいはZDV+3TCと本剤を併用した臨床試験期間中において、ウイルス量の著しい再上昇(リバウンド)を経験した患者から分離されたウイルス分離株において、K103N置換は最も頻繁に認められたRT変異であった。RTの100、101、108、138、188又は190番目のアミノ酸置換も認められたが、より少ない頻度であり、K103N置換を伴った場合にしか認められないことが多かった。本剤投与前の患者から得られた検体中には、K103N置換は認められなかった。本剤に耐性を示すRTのアミノ酸置換様式は、本剤と併用投与された他の抗ウイルス療法剤とは関係していない。
18.4 他の抗ウイルス薬に対する交差耐性
本剤、ネビラピン及びデラビルジンに対する細胞培養での交差耐性プロフィールは、K103N置換が3種すべてのNNRTIsに対する感受性を損失させることを示していた。試験したデラビルジン耐性臨床分離株3株のうち2株は、本薬に対する交差耐性であり、かつ、K103N置換を含んでいた。残りの1株は、RTの236番目のアミノ酸置換を持ち、本剤とは交差耐性を示さなかった。
本剤の臨床治験症例で治療不良患者(ウイルス量のリバウンド)の末梢血単核細胞から得られたウイルス分離株のNNRTIsに対する感受性を評価した。本剤に耐性であることがあらかじめ確認された13種の臨床分離株は、ネビラピン及びデラビルジンに対しても耐性であった。これらのNNRTIs耐性分離株の5株にはRTのK103N、あるいは108番目のバリンイソロイシン置換(V108I)が認められた。試験した本剤治療不良分離株の3株には、細胞培養中での本剤に対する感受性が残っており、ネビラピン及びデラビルジンに対しても感受性であった。
本剤とPIは標的酵素が異なるために、両薬剤間の交差耐性の可能性は低い。
本剤とNRTIとの交差耐性は標的結合部位と作用機序が異なるので、その可能性は低い。
19. 有効成分に関する理化学的知見
| 一般名 | エファビレンツ(Efavirenz) |
|---|---|
| 化学名 | (-)-(S)-6-Chloro-4-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H -3,1-benzoxazin-2-one |
| 分子式 | C14H9ClF3NO2 |
| 分子量 | 315.67 |
| 融点 | 130~136℃ |
| 性状 | 白色~微帯赤白色の粉末である。メタノール又はエタノール(99.5)に溶けやすく、水にほとんど溶けない。 |
| 構造式 | 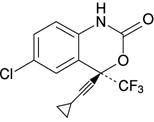 |
22. 包装
〈ストックリン®錠200mg〉
90錠[瓶、バラ]
〈ストックリン®錠600mg〉
30錠[瓶、バラ]
23. 主要文献
- Schneider S, et al. J Acquir Immune Defic Syndr. 2008; 48: 450-4.
- Haas DW, et al. AIDS. 2004; 18: 2391-400.
- The International HapMap Consortium. Nature. 2007; 449: 851-61.
24. 文献請求先及び問い合わせ先
MSD株式会社 MSDカスタマーサポートセンター
東京都千代田区九段北1-13-12
医療関係者の方:フリーダイヤル0120-024-961
26. 製造販売業者等
26.1 製造販売元
MSD株式会社
東京都千代田区九段北1-13-12